TPHD KEMİK İLİĞİ YETMEZLİKLERİ OKULU LALE OLCAY
|
|
|
- Tülay Baydar
- 7 yıl önce
- İzleme sayısı:
Transkript
1 TPHD KEMİK İLİĞİ YETMEZLİKLERİ OKULU LALE OLCAY 15 KASIM 2014

2 DERS AKIŞI Diskeratosis kongenita Mitokondrial Hastalıklar *Pearson sendromu *Retiküler disgenesis Kongenital trombositopeniler *Amegakaryositik trombositopeni *TAR sendromu Kongenital diseritropoietik anemi

3 DİSKERATOSİS KONGENİTA Zinsser-Cole-Engman sendromu

4 DİSKERATOSİS KONGENİTA Diğer adı: Zinsser-Cole-Engman sendromu X-e bağlı, otozomal dominant, otosomal resesif geçebilen, ancak hastaların çoğunda sporadik olan bir kemik iliği yetmezlik sendromudur Kalıtsal aplastik anemi şüphe edilen hastalarda Fankoni anemisinden sonra dışlanması gereken sendromdur Klasik özellikleri *Mukokutanöz üçlü (ciltte pigmentasyon, distrofik tırnak, mukozal lökoplaki) görülmesi *Telomerlerin kısa olması *İleri dönemde kemik iliği yetmezliği ve malignensi (skuamöz hücreli karsinom) görülmesidir *Hastaların %75 inde bir fiziksel anomali vardır
 görülmesidir *Hastaların %75 inde bir")
5 DİSKERATOSİS KONGENİTA: TANI YAŞI, CİNS VE BELİRTİLERİN YAŞ İLE İLİŞKİSİ Erkek/Kız: 3.2/1 Median tanı yaşı: 14 (doğum sırası- 75 yaş) Hoyeraal-Hreidarsson (HH) ve Revesz sendromu (RS) olanlarda çok erken yaşta tespit edilir Atipik DK olguları geç yaşlarda tespit edilir
 ve Revesz sendromu (RS) olanlarda çok")
6 DİSKERATOSİS KONGENİTA: TANI YAŞI Shimamura Blood rev 2010;24:

7 DİSKERATOSİS KONGENİTA: PATOGENEZ MODELİ
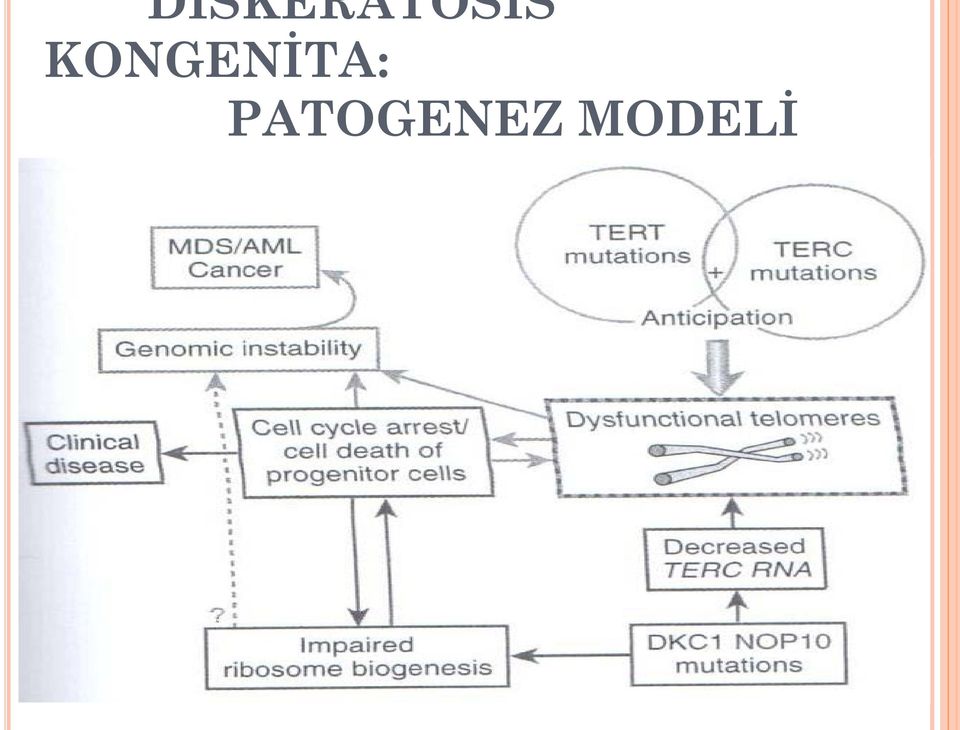
8 TELOMER VE TELOMERAZ Telomerler, lineer kromozomların sonlarında yer alır. Kromozom sonlarını parçalanmaktan ve uç uca füzyon yapmaktan korur Hekzanükleotid (TTAGGG) tekrarları+ilgili proteinler= şelterin kompleksi Hücre çoğaldıkça (ve hücre yaşlandıkça) telomerler kısalır Telomerler çok kısaldığı zaman, hücre siklusunda arrest olur veya hücre ölümü gerçekleşir Telomeraz enzimi (ribonükleoprotein kompleksi), progenitör hücrelerin telomerlerinin uzatılması işinde görev görür Telomeraz: Başlıca reverse transkriptaz TERT + RNA molekülü TERC. Telomer tekrarlarının sentezi için bir kalıp (template) görevi görür DK da, telomeraz fonksiyonu yetersizdir; en fazla, en hızlı bölünen hücreler (mukokutanöz ve hematopoietik hücreler) etkilenir

9 OSKİ Telomerlerin yapısı. Telomerler, kromozomların sonunda yer alır. Kromozom sonlarını parçalanmaktan ve uç uca füzyon yapmaktan koruyan protein DNA kompleksidir. Bölünmeyen hücrelerde telomerler, ilmek şeklindedir ve bu şekilde DNA tamir proteinleri ve DNA ekzonükleazları, kromozom ucuna ulaşamaz Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,

10 TELOMERAZIN GÖREV YAPMA ŞEKLİ Telomeraz, telomerlerin 3 sonuna telomerik tekrarlar eklemektedir

11 TELOMER-TELOMERAZ BİYOLOJİSİ Telomer biyolojisi. Lineer kromozomların sonlarında yer alan telomerler, şelterin kompleksi denilen, hekzanükleotid (TTAGGG) tekrarları + ilgili proteinlerden oluşur. Telomeraz kompleksi, hücre çoğaldıkça telomerler kısaldığı için, telomerlerin sonuna telomerik tekrarlar ekleyerek telomerlerin uzatılmasından sorumludur. DK genlerinin altı tanesi telomeraz kompleksinin bir parçasını oluşturan proteinleri kodlar (Diskerin, TERT, TERC, NHP2, NOP10, WDR79/TCAB1). Yedinci gen, TINF2, şelterin kompleksinin bir parçası olan TIN2 proteinini kodlar.parikh Curr Opin Pediatr 2012; 24: 23-32
.")
12 DİSKERATOSİS KONGENİTA GENLERİ DK genlerinin altısı telomeraz kompleksinin bir parçasını oluşturan proteinleri kodlar (Diskerin, TERT, TERC, NHP2, NOP10, WDR79/TCAB1). Yedinci gen, TINF2, şelterin kompleksinin bir parçası olan TIN2 proteinini kodlar

13 DİSKERATOSİS KONGENİTA DA GÖRÜLEN MUTASYONLAR Telomer i idame ettirmede işlevi olan 7 geni mutasyonu bilinmekte. Parikh ve ark. Curr Opin Pediatr 2012; 24: Gen Kromoz om lokusu Kalıtım şekli Sıklık Kodladığı protein DKC1 Xq28 XLR, de novo %30 Diskerin i kodlar TINF2 14q11.2 Otozomal dom, de novo %15 TIN2 i kodlar TERC 3q26.3 Otozomal dom %10 Aynı protein TERT 5p15.53 Otozomal dom, otozomal res, de novo NOP10/NOLA3 15q14- q15 %5-10 Aynı protein Otozomal resesif %1 Aynı protein NHP2/NOLA2 5q35.5 Otozomal resesif %1 Aynı protein TCAB1/WDR79/ WRP53 17p13.1 Otozomal resesif 2 olgu Aynı protein

14 TELOMER BİYOLOJİSİ VE DK DA GÖRÜLEN MUTASYONLAR Telomerler, kromozomların sonunda sarı noktalar olarak gösteriliyor. Telomerler, hücre çoğalması sırasında eklenen TTAGGG tekrarlarıdır. TERT, telomeraz enzimi dominanttır. TERC, RNA kalıbıdır ve hem dominant, hem resesifdir. DKC1, X-e bağlı resesif gen olup diskerin i kodlar. NOP10/NOLA3 ve NHP2/NOLA2 otozomal resesiftir. TINF2, şelterin i idame ettiren TIN2 i kodlar. Shimanura Blood Rev 2010; 24:

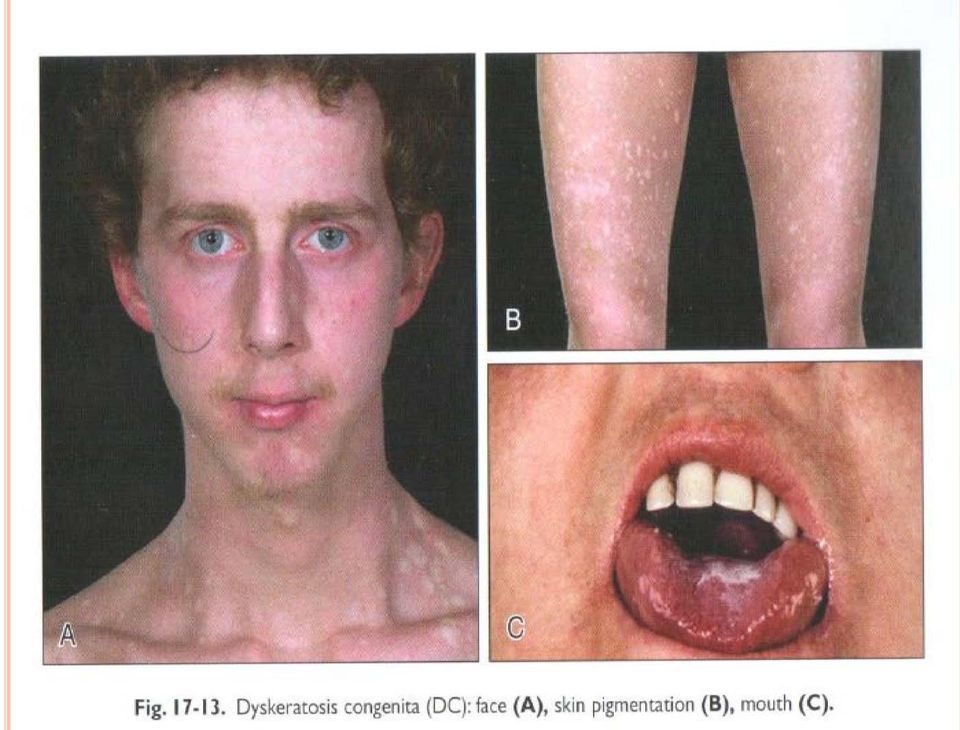
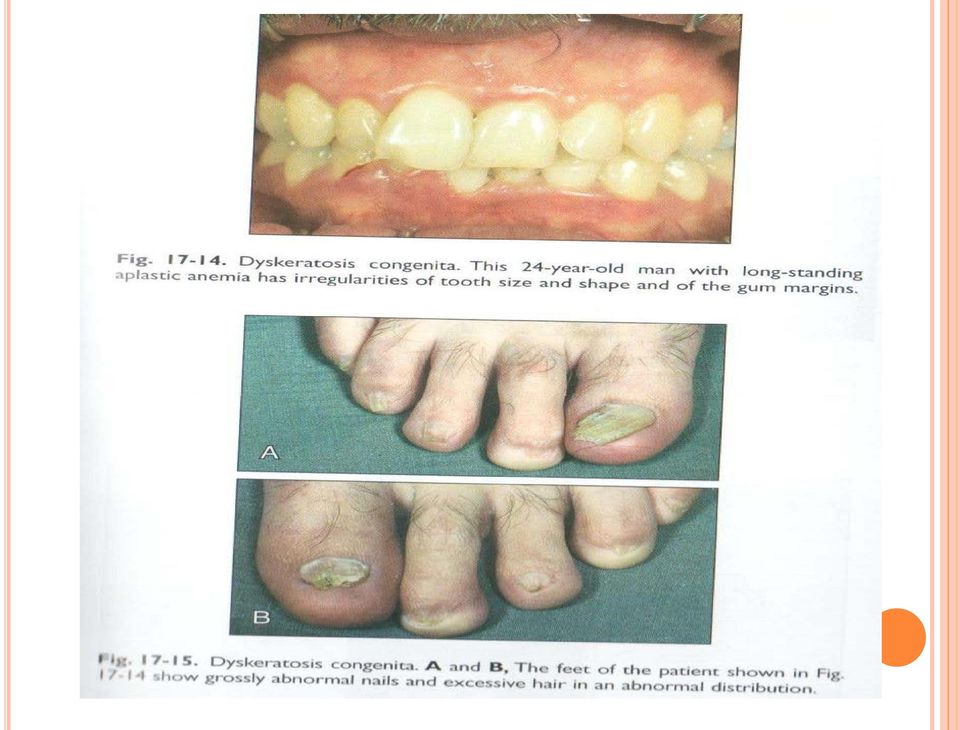
15 DİSKERATOSİS KONGENİTA DA BAŞLICA BELİRTİ VE BULGULAR-1 Tanısal üçlü: *Tırnak distrofisi: %70 *Ciltte dantelsi retiküler pigmentasyon: %67, *Oral lökoplaki: %47 Gözler: %29. Gözyaşı kanalında stenoz, epifora, blefarit, eksudatif retinopati, vasküler retinopati, strabismus, katarakt, kirpik yokluğu, ülser Saç: %19. Seyrek, ince, alopesi, erken kayıp, erken beyazlama, seyrek kaş Dişler: %13. Çürük, kayıp, anormal şekil, periodontit, gövde (crown)/kök oranında azalma, taurodontizm Gelişme: %13. Gelişme geriliği Gastrointestinal: %12. özafagus darlığı, striktür, ağ (%8); karaciğer sirozu, fibroz, karaciğer bozukluğu (%2) Boy kısalığı: %12 İskelet: %10. Osteoporoz (%6), kalçada avasküler, nekroz Kafa: Mikrosefali (%9), serebeller hipoplazi (%5), intrakranial kalsifikasyon (%3), Dandy-Walker, tonsiller herniasyon Düşük doğum ağırlığı (%9) Kardiyopulmoner (%7): Pulmoner fibrosis, perfüzyonda azalma, restriktif akciğer hastalığı, arterio-venöz fistül Kardiyak (%1): ASD, VSD, dilate kardiyomiyopati Genitoüriner (%7): Fimosis, meatal stenoz, üretral striktür, hipospadias, penil
; karaciğer sirozu, fibroz, karaciğer bozukluğu (%2) Boy kısalığı: %12 İskelet: %10.")
16 DİSKERATOSİS KONGENİTA: BAŞLICA BELİRTİ VE BULGULAR-2 Gonadlar: %3. Erkek: Küçük testis, inmemiş testis Kız: Atrofi, konstriksiyon, vajinal lökoplaki Hiperhidrosis (%6) Hoyeraal-Hreidarsson sendromu (HH) (%5) Sistemik DK + serebeller hipoplazi + gelişme bozukluğu Revesz sendromu(%4): (RS) HH + eksudatif retinopati Nörolojik bulgular (%4): Ataksi, spastisite, hipotoni Nöropsikiyatrik (%25, %83): Dikkat eksikliğihiperaktivite bozukluğu, anksiyete, ruh hali bozuklukları, gelişme bozukluğu Kulaklar (%2): Sağır, dönük, düşük kulak Shimamura ve ark. Blood Rev 2010;24: Chirnomas ve ark. Pediatr Clin N Am 2013; 60:
: Dikkat eksikliğihiperaktivite bozukluğu, anksiyete, ruh hali bozuklukları, gelişme bozukluğu Kulaklar (%2): Sağır, dönük, düşük")
17
18
19 KLİNİK BULGULARA GÖRE SINIFLANDIRMA Klasik DK Sessiz taşıyıcılar Atipik DK

20 KLİNİK BULGULARA GÖRE SINIFLANDIRMA : KLASİK DK Tipik olarak, hastalar doğduklarında normaldir Mukokutanöz bulgular çocukluk döneminde ortaya çıkar Kemik iliği yetmezliği ergenlik döneminde ortaya çıkar Hastalar 3. dekadda kaybedilir

21 KLİNİK BULGULARA GÖRE SINIFLANDIRMA: SESSİZ TAŞIYICILIK *Hasta bir DK olgusunun aile bireyleri arasındadır *Bu belirti ve bulgulardan hiçbirine sahip değildirler *Ancak telomerleri çok kısadır *Yakın izlemleri gerekir
22 DİSKERATOSİS KONGENİTA: KLİNİK BULGULARA GÖRE SINIFLANDIRMA: ATİPİK DK Şu şekilde karşımıza çıkabilir: *Sadece edinsel aplastik anemi var, mukokutanöz bulgular yok *DK bulguları var; mukokutanöz bulgular hariç *DK bulguları var; hematolojik bulgular hariç *Sadece pulmoner fibrosis var
23 A.TERC mutasyonu saptanmış olan DK lı bir ailenin aile ağacı B.Anne ve annenin kızkardeşinin TERC için heterozigot olup, kız çocuğun bu mutasyonu taşımadığını gösteren kromatogram C.Annenin telomer boyunun 1 persentlin altında olduğu, ancak etkilenmemiş olan kız çocuğun telomer boyunun normal olduğunu gösteren
24 DİSKERATOSİS KONGENİTA: KOMPLİKASYONLAR: MALİGN HASTALIK GELİŞİMİ *Üçüncü dekadda gelişir; hastalığın hafif ve geç başlangıçlı tiplerinde daha sıktır. *Türleri Solid Tümörler (%20-30) *Skuamöz hücreli karsinom Baş, boyun: Dil, dudak, ağız, damak,yanak,nazofarinks, larinks Üst-alt GIS: özafagus, anüs Kadınlarda genital bölge *Cilt kanseri *Adenokarsinom (mide, kolon, akciğer, pankreas) MDS (%30) AML (%10) Atipik DK ve geç başlangıçlı formlarda AML daha sık; en sık rastlanan kromozom anormalliği 7.kromozom anormallikleri 1) Shimamura ve ark. Blood Rev 2010;24: ) Bessler M ve ark. Nathan and
25 DİSKERATOSİS KONGENİTA: LABORATUVAR BULGULARI 1) Periferik kan bulguları 2) Kemik iliği incelemesi 3) İmmünolojik Bulgular 4) Sitogenetik (Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009, )
26 DİSKERATOSİS KONGENİTA: PERİFERİK KAN BULGULARI Pansitopeni: Hastalarda aplastik anemi gelişme riski 20 yaşa kadar %1 iken, sonraki her dekatta yaklaşık %10 artar (50 yaş civarında kümülatif insidans: yaklaşık %50) 1 Başka kaynaklara göre 40 yaşta, hastaların %95 inden fazlasında pansitopeni gelişmiş olur 2 Sitopeni (bir veya birden fazla seride) (%80) Makrositoz (anemi ile birlikte veya değil) Trombositopeni Nötropeni Dolaşan progenitör hücrelerde azalma Hemoglobin F de artma Von Willebrand faktörde artma 1) Shimamura ve ark. Blood Rev 2010;24: ) Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı,
27 DİSKERATOSİS KONGENİTA DA KEMİK İLİĞİ BULGULARI Hiposellüler kemik iliği (3 seride birden hipoplazi) Mast hücre sayısında artış Diseritropoez Hiposellüler MDS MDS/AML bulguları Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
28 DİSKERATOSİS KONGENİTA DA İMMÜNOLOJİK BULGULAR Humoral anormallikler Hipo-hiper gama globulinemi Sellüler anormallikler Lenfopeni : B, T, NK Fonksiyonel anormallikler Uyarı ile proliferasyonda azalma Uyarı ile apoptozda artma Anerji EBV transformasyonu ile immortalizasyon güçlüğü (X-e bağlı olan DK için) Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
29 DİSKERATOSİS KONGENİTA DA SİTOGENETİK Sitogenetik instabilite: kültürü yapılmış hücrelerde klonal olmayan kompleks anomalileri Kromozom kırıkları, hipodiploidi, mikronukleus Anafaz ve telofaz köprüleri* Kısa telomer sinyalsiz uç* Telomer sinyali olmadan uç uca füzyon* DNA çapraz bağlayıcı ajanların klastojenik etkisi ile kromozom instabilitesinde artış yok Kİ hücrelerinde klonal anormallikler: monozomi 7 (MDS, MDS/AML) Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
30 DİSKERATOSİS KONGENİTA DA SELLÜLER ANORMALLİKLER Spontan kromozom kırıkları Çapraz bağlayıcı ajanlarla (DEB, mitomisin C, nitrojen mustard gibi) kromozom kırıklarında artış olmaması İyonize radyasyona hipersensitivite (fikir birliği yok) Belirli sitotoksik ajanlara karşı hipersensitivite Kısa telomer* Prematür replikatif yaşlanma* TERT ile mortalizasyonun yapılamaması (Xe bağlı) Taraflı X sessizleştirilmesi (X-e bağlı DK nın hanım taşıyıcılarında) Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
31 DİSKERATOSİS KONGENİTA DA TELOMER BOYU Telomer kısalığı (yaş için normalin %1 inin altı) (şekil) Çok renkli akım floresans in situ hibridizasyon yöntemi ile belirlenen grafikler, DK ı, Fankoni anemisi, etkilenmemiş akrabalar ve DK olmayan kalıtsal kemik iliği yetmezliğinden ayırdetmeye yarar Sensitivite ve spesifitesi yüksektir Halen, çeşitli lökosit alt gruplarında çok kısa telomer varlığı, DK tanısı için, en kullanışlı tarama testidir
32 DİSKERATOSİS KONGENİTA DA TELOMER BOYU DK hastaları ve akrabalarında, çok renkli akım floresans in situ hibridizasyon yöntemi ile ölçülmüş olan yaşa göre telomer boyu. Vertikal eksen, telomer boyunu (kilobayt) göstermekte. Eğriler 400 normal kontrol bireyine göre belirlenen , 50., 90., 99. persentilleri gösteriyor. DK hastası n:17; HH varyantı n:4; RS n:4; seessiz taşıyıcı n:1; akrabalar n:54; Alter ve ark. Blood 2007;
33 DK dışı hastalar ve akrabalarında yaşa bağlı telomer uzunluğu. Vertikal eksen, telomer boyunu (kilobayt) göstermekte. Eğriler 400 normal kontrol bireyine göre belirlenen , 50., 90., 99. persentilleri gösteriyor. Fankoni anemisi (FA) n: 13; FA -KİT sonrası n:1; FA mozaisizm n:3; Diamond Blackfan anemisi n: 14; Shwachman Diamond sendromu n:5; diğer n:10; akrabalar n: 35. Alter ve ark. Blood 2007; 110:
34 DİSKERATOSİS KONGENİTA DA MUTASYON CİNSİ İLE HASTALIĞIN CİDDİYETİNİN İLİŞKİSİ Gen Kromozo m lokusu Kalıtım şekli Sıklı k Ciddiyet DKC1 Xq28 XLR, de novo %30 Çok hafif-çok ağır TERC 3q26.3 Otozomal dom TERT 5p15.53 Otozomal dom, otozomal res, de novo %10 Hafif. Atipik DK, geç başlangıç; AC fibrosisi, siroz, hiposellüler MDS- MDS/AML Genetik antisipasyon görülür. Tam olmayan (İnkomplet) penetrans %5-10 Atipik DK, AC fibrosisi, aplastik anemi, genetik antisipasyon.tam olmayan (İnkomplet) penetrans Biallelic TERT bir olguda, Bir de HH sendromunda Biallelik TERT veya TERC null mutasyonu tarif
35 DİSKERATOSİS KONGENİTA DA PROGNOZ Ölüm nedenleri: Kemik iliği yetmezliği>immün yetmezlik >pulmoner komplikasyonlar>malign hastalıklar
36 DİSKERATOSİS KONGENİTA DA HASTALIK ŞİDDETİNİN ÖNGÖRÜLMESİ : HASTALIK (GENETİK) ANTİSİPASYONU Genetik antisipasyon: Nesilden nesile geçen bir hastalık, her yeni nesilde, bir öncekinden daha erken yaşta ve bir öncekinden daha ciddi olarak karşımıza çıkar. Otozomal dominant DK da hastalık antisipasyonu görülür; nesilden nesile aktarılan telomerlerin giderek kısalması nedeniyle gelişir Shimamura ve ark. Blood Rev 2010;24:
37 DK DA VARYANTLAR Hoyeraal-Hreidarsson (HH) sendromu DK+serebeller hipoplazi+gelişme bozukluğu Revesz sendromu (RS) HH + eksudatif retinopati
38 HOYERAAL-HREİDARSSON (HH) SENDROMU Erken çocukluk döneminde ortaya çıkar İntrauterin büyüme geriliği, mikrosefali, serebeller hipoplazi, mental retardasyon, ilerleyici kombine immün yetmezlik, aplastik anemi Miyelinizasyonda gecikme, korpus kallozumda hipoplazi görülür Çoğunda DKC1 geninde mutasyon vardır Homozigot TERT, bir olguda Heterozigot TERT +genetik antisipasyon olgusu..1 adet Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
39 HOYERAAL-HREİDARSSON (HH) SENDROMU Hoyeraal-Hreidarsson (HH) sendromu (%5): Sistemik DK + serebeller hipoplazi + gelişme bozukluğu Shimamura Blood rev 2010;24: , Chirnomas ve ark Pediatr Clin N Am 2013; 60:
40 REVESZ SENDROMU İlerleyici bilateral eksudatif retinopati (Coat s retinopathy) İntra uterin büyüme geriliği İnce, seyrek saç İnce retiküler cilt pigmentasyonu Serebeller hipoplaziye bağlı ataksi Serebral kalsifikasyonlar Ekstensör hipertoni İlerleyici psikomotor gerilik İlerleyici kemik iliği yetmezliği Erken çocuklukta ölüm A R282H TIN2 mutasyonu: bir olgu Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
41 ATAKSİ-PANSİTOPENİ SENDROMU Kemik iliği yetmezliği + ciddi serebeller hipoplazi + kromozomal instabilite DEB/Mitomisin C ile kromozom kırıklarında artış yok MDS, MDS/AML ve monozomi 7 birlikte görülebilir Moleküler yapısı ve etyolojide telomerlerin yeri incelenme aşamasında Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
42 DK DA TEDAVİ-1 KEMİK İLİĞİ YETMEZLİĞİ Klasik DK da kemik iliği yetmezliği, Fankoni anemisi gibi tedavi edilir. Androjen ve hematopoietik büyüme faktörleri yetmezliği düzeltebilir; fakat hastaların çoğunda etki geçicidir Androjen FA e göre daha düşük dozda verilir (oksimetholon mg/kg/g) EPO + G-CSF birlikte etkilidir Androjenlerle büyüme faktörleri aynı anda kullanılmamalı (splenik peliosis ve splenik rüptür riski nedeni ile) İdiopatik aplastik anemide kullanılan ATG ve siklosporin etkisizdir Shimamura ve ark. Blood Rev 2010;24: Bessler M ve ark. Nathan and Oski s Hematology of
43 DK DA TEDAVİ-1 KEMİK İLİĞİ YETMEZLİĞİ-(DEVAM) Hematolojik anormallik yok ise veya kemik iliği yetmezliği belirtileri hafif ise, sadece hemogram değerleri izlenir Hematolojik anormalliği olan hastalara, yıllık kemik iliği aspirasyonu ve biyopsisi yapılır. MDS morfolojisi ve sitogenetiği incelenir. Muhtemel HKHN için plan yapılır (hematolojik komplikasyonların gelişimi hızlı olabilir) DK zemininde gelişen MDS, AML nin tedavisi için bir standart yoktur Geçici olarak klonal sitogenetik anormallikler gelişebilir. Persistan veya kompleks ise kötü prognoz göstergesidir Shimamura ve ark. Blood Rev 2010;24: Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
44 DK DA TEDAVİ-2 Hematopoietik kök hücre nakli (HKHN), en son tedavi seçeneğidir Ancak mortalite ve morbiditesi yüksektir Düşük intensiteli hazırlama rejimi, radyoterapi içermeyen ve bleomisin olmayan rejimler kullanılır Beklenmeyen komplikasyonlar: Hepatik, pulmoner HKHN yapılan hastalarda median yaşam süresi: 7 yıl HLA tam uyumlu kardeşten yapılan nakillerde 5 yıllık yaşam %68, alternatif donörlerden yapılan nakillerde %31 Shimamura ve ark. Blood Rev 2010;24: Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
45 DK DA TEDAVİ-3 PULMONER HASTALIKLAR Pulmoner hastalığın tedavisi yoktur İmmünosupresiflere yanıt vermez Akciğer transplantasyonu düşünülebilir DK hastaları -sigara içmemeli -pulmoner toksisite yapan ilaçlardan (bleomisin gibi) uzak durmalı -kanser tedavisi ve KİT sırasında akciğerleri korunmalı Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
46 DK DA TEDAVİ-4 MALİGN HASTALIKLAR Ultraviyole ışınlarından korunmalı, güneş kremi sürmeliler Özellikle oral lökoplakisi olanlar sigara ve alkol kullanmamalı Düzenli Diş Hekimi tarafından kontrol edilmeliler Malignensi taramaları yapılmalı (özellikle GIS) Kemik iliği yetmezliği nedeni ile, kemoterapi ve cerrahi müdahale sınırlı olarak uygulanabilir Oral malignensilerde premalign hiperkeratotik lezyon düzenli takip edilir ve biyopsisi alınır Neoadjuvan radyoterapi veya belirlenmiş olan malign lezyonların cerrahi rezeksiyonu, ipsilateral lenf nodlarının temizlenmesi esastır. Adjuvan radyoterapi eklenebilir veya eklenmez Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
47 KARTİLAJ-SAÇ HİPOPLAZİSİ (METAFİZİYEL KONDROPLAZİ)
48 KARTİLAJ-SAÇ HİPOPLAZİSİ=METAFİZİYEL KONDROPLAZİ Otozomal resesif kondroplazi Kısa boy (ekstremite kısalığı) Saçlar kısa, seyrek ve merkezi pigmentini içermiyor Göğüs deformitesi Alt ekstremitelerde varus Lordoz Skolyoz Agangriyonik megakolon (Hirschsprung hastalığı)
49 KARTİLAJ-SAÇ HİPOPLAZİSİ=METAFİZİYEL KONDROPLAZİ HEMATOLOJİK BULGULAR *Hafif-ciddi makrositik anemi: Çoğunluğunda var *Lenfopeni: Çoğunluğunda var *Nötropeni: 1/4 ünde var
50 KARTİLAJ-SAÇ HİPOPLAZİSİ: ANEMİ Hafif, erken çocukluk döneminde kendiliğinden düzelir Ciddi olgularda: DBA e benzer Ciddi immün yetmezlik ile birlikte olabilir Anemi ciddiyeti Boy ile ters ilişkili İmmün yetmezlik ile doğrusal ilişkili Lenfoid malignansi ve bazal hücreli karsinom, normal popülasyondan 7 kat fazla
51 KARTİLAJ-SAÇ HİPOPLAZİSİ : AYIRICI TANI Diamond Blackfan Anemisi (DBA) Trombositoz, EPO yüksekliği, eritrosit ADA sında artış nedeni ile karışır DBA de metafiziyel displazi yoktur Mutasyonlar farklıdır
52 KARTİLAJ-SAÇ HİPOPLAZİSİ: GENETİK Otozomal resesif geçişli RMRP nin RNA sını kodlayan gende mutasyon RMRP, mtdna replikasyonu, ribozom biyogenezi ve pre-rna processing i, siklin B2 nin mrna sının parçalanması ve bu şekilde hücre siklusunun ilerlemesinde işlev görür Ribozomal birleşme sırasında rrna kesilmesinde (bölünme) azalma ve kemik displazisinin derecesi arasında ciddi bir doğrusal ilişki rrna kesilmesinde (bölünme) azalma ve hücre siklusu bozukluğu ile saç hipoplazisi, immün yetmezlik, hematolojik anormallik ve kanser riski artışı arasında ilişki vardır
53 KARTİLAJ-SAÇ HİPOPLAZİSİ: TEDAVİ Transfüzyon Steroid tedavisi: İmmün yetmezlik ve boy kısalığı yapıcı etkileri nedeni ile dikkatle verilmeli HKHN: Kemik iliği yetmezliği, hastaların çoğunda geçer, fakat bazı olgularda ciddi ve inatçıdır Ciddi kombine immün yetmezliği olan olgularda miyeloablazyon ile birlikte olarak veya olmayarak
54 MİTOKONDRİ HASTALIKLARI PEARSON SENDROMU
55 PEARSON SENDROMU Pearson un kemik iliği-pankreas sendromu Tipik özellikleri: *Refrakter sideroblastik anemi *Kemik iliği öncüllerinde vakuolizasyon *Mitokondrial DNA (mt DNA) da delesyon
56 PEARSON SENDROMU: PATOGENEZ *Mitokondrial DNA daki delesyon veya dublikasyonlardan kaynaklanır *Mitokondriler, hem nukleus, hem de mitokondri DNA sı tarafından kontrol edilir *Mitokondri DNA sı solunum yolu enzimlerinin 13 tanesini ve mitokondride protein sentezi için gerekli olan 24 RNA molekülünü kodlar *Oksidatif fosforilasyon (solunum yolu enzimlerin yetersizliği nedeni ile) bozukluğu sonunda NAD, sitokrom oksidaz, ATP, trna ve rrna azalır *Hastalık ciddiyeti, mutant/vahşi mtdna oranına ve mutant mtdna nın dokulardaki dağılımına bağlıdır
57 MİTOKONDRİ DNA SININ DİĞER ÖZELLİKLERİ *Genellikle maternal yolla kalıtılır *Pearson sendromunda, mtdna daki mutasyon, heteroplazmik tir. *Heteroplazmik mutasyon: Mutasyon, bir hücredeki mt genomların ancak bazılarında vardır ve bu nedenle gebelik sırasında bebeğin etkilenme derecesini önceden belirlemek zordur
58 PEARSON SENDROMU: KLİNİK BULGULAR Erken çocukluk döneminde başlar (ilk 6 ay); K/E:0.7/1, tüm etnik gruplarda görülür Fiziksel bir anomali, nadir olarak bulunur Pansitopeni Ekzokrin pankreas bozukluğu İnsuline bağlı diabetes mellitus Karaciğer yetmezliği Böbreklerde tubulopati Laktik asidoz Hematopoietik öncüllerde vakuolizasyon Hidrops fetalis Chirnomas ve ark. Pediatr Clin N Am 2013;60: Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
59 PEARSON SENDROMU: HEMATOLOJİK BULGULAR *Anemi, genellikle makrositiktir *Nötropeni ve trombositopeni (hastaların ½ sinde) *Miyeloid ve eritroid öncüllerde vakuolizasyon *Eritroid seride azalma *Halka eritroblast varlığı hemen hemen tüm hastalarda
60 PEARSON SENDROMU: PROGNOZ Hastaların çoğu, 3 yaşından önce ölür, median yaşam süresi (projected) 4 yıl, 10 yaşında %36 Başlıca ölüm nedeni, sırasıyla metabolik asidoz, karaciğer ve böbrek yetmezliğidir Az sayıda hasta hematolojik düzelme gösterir; fakat sonunda Kearns-Sayre sendromu gelişir Malign transformasyon bildirilmemiş
61 PEARSON SENDROMU
62 PEARSON SENDROMU: TEDAVİ Anemi: Eritrosit suspansiyonu transfüzyonu EPO Nötropeni: G-CSF
63 MİTOKONDRİ HASTALIKLARI RETİKÜLER DİSGENESİS
64 RETİKÜLER DİSGENESİS: PATOGENEZ Mitokondride bulunan adenilat kinaz 2 geninde mutasyon var Katlanmamış protein cevabı yok Sekretuar protein yükünde dengesizlik
65 RETİKÜLER DİSGENESİS: KLİNİK BULGULAR Neonatal dönemde başlar Lökopeni ve lenfopeni + Kemik iliğinde miyeloid ve lenfoid öncüller yok ancak kemik iliğindeki aplazi tam değil Lenfoid doku genellikle yok Bazı hastalarda anemi ve trombositopeni de var
66 RETİKÜLER DİSGENESİS: TEDAVİ Ciddi kombine immün yetmezlik gibi karşımıza çıkan olgularda hematopoietik kök hücre nakli ile kür sağlanmıştır Mortalite yüksektir
67 KALITSAL TROMBOSİTOPENİLER AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT)
68 AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT) Bebeklik döneminde başlayan İzole trombositopeni Kemik iliğinde megakaryosit azlığı veya yokluğu Fiziksel anomali yokluğu ile seyreden Otozomal resesif geçişli Trombopoietin reseptör mutasyonu (protoonkogen MPL) na bağlı olan İzole trombositopeninin, çoğunlukla aplastik anemiye dönüştüğü kalıtsal bir kemik iliği yetmezliğidir
69 AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT): KLİNİK BULGULAR Doğumdan sonraki ilk haftada peteşi ve kanama bulguları Ciddi trombositopeni Trombosit morfoloji ve boyu normal Kemik iliği incelemesi: Megakaryosit yok veya az Trombopoietin yüksek Fiziksel anomali genellikle yok; ancak eğer varsa özgün değil Psikomotor gerilik bulunabilir Daha az ciddi biçiminde: Trombositopeni orta dereceli veya geçici
70 AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT): PROGNOZ CAMT, ilk 10 yılda ağırlaşır ve ikinci dekadda ciddi aplastik anemiye döner MDS veya MDS/AML e dönüşüm olabilir
71 AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT) AYIRICI TANI: KALITSAL HASTALIKLAR 1) Radius yokluğu ile birlikte olan trombositopeni (TAR): TAR da görülen ön kol anomalisi (radius yokluğu) CAMT da yoktur 2) Wiskott Aldrich sendromu: WAS daki mikro trombositler, CAMT da yoktur 3) Noonan Sendromu: Noonan sendromunda var olan yüz ve kalp anomalileri, PTPN11 mutasyonu CAMT da yoktur Aplastik anemiye dönüştükten sonra 1) Fankoni anemisi: Fankoni anemisindeki DEB ve MMC test pozitifliği CAMT da yoktur 2) Diskeratosis kongenita:
72 AYIRICI TANI:EDİNSEL HASTALIKLAR: TROMBOSİT YIKIMININ ARTTIĞI DURUMLAR İMMÜN NEDENLERE BAĞLI *Neonatal alloimmün trombositopeni *Annedeki İTP *İlaca bağlı antikor *Otoimmün trombositopenik purpura İMMÜN OLMAYAN NEDENLERE BAĞLI OLARAK *Psödotrombositopeni *DİK *Asfiksi *Perinatal aspirasyon *NEK *Kasabach-Meritt sendromu *Neonatal tromboz *Respiratuvar distres sendromu *Annede preeklampsi *Kardiyopulmoner bypass (ECMO) -Ailevi trombotik trombositopenik purpura DİĞER NEDENLERLE *Hiperbilirübinemi *Fototerapi *Polisitemi *Rh hemolitik hastalığı *Total parenteral nutrisyon Cantor A. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
73 AYIRICI TANI:EDİNSEL HASTALIKLAR 2 TROMBOSİT YAPIMININ AZALDIĞI DURUMLAR Viral hastalıklar (HIV, CMV, Rubella, HHV- 6) SEKESTRASYON Hipersplenizm Kongenital lösemi Down sendromunda geçici miyeloproliferatif hastalık Nöroblastom Histiyositoz Osteopetroz Cantor A. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009,
74 AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT): GENETİK Otozomal resesif geçişlidir Trombopoietin reseptör geni olan c-mpl de çeşitli mutasyonlar (1p34 de yer alır) Hastalar homozigot veya çift heterozigottur Genotip-Fenotip İlişkisi Tip I: c-mpl de tam fonksiyonel yokluk Tip II: c-mpl de az miktarda fonksiyon varlığı: Trombosit sayısında geçici düzelme ve pansitopeniye dönüşümde Tip I e göre gecikme vardır. Pansitopeni, ilk belirti olabilir AA, AML, MDS dönüşümün, özgün olarak genetik mutasyon ile ilişkisi olup olmadığı bilinmiyor Taşıyıcılar (heterozigot) Trombosit sayısı normal olabilir, ancak megakaryopoiesis bozuktur Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009, ; Shimamura ve ark. Blood Rev 2010;24:
75 AMEGAKARYOSİTİK TROMBOSİTOPENİ (CAMT): TEDAVİ Trombosit suspansiyonu transfüzyonu (ışınlı, filtreli) DDAVP: Büyük çocuklarda kullanılabilir Büyüme faktörleri ve sitokinlerle (IL-3, GM-CSF) uzun süreli yanıt alınmaz HKHN, küratif olan tek tedavi seçeneğidir *c-mlp mutasyonu için heterozigot olan kardeşler başarı ile kullanılmıştır *Akraba olmayan vericilerden yapılan transplantların başarısı düşüktür (toksisite ve engraftman başarısızlığı) İleride gelişecek gen tedavisinden yararlanacağı düşünülmekte Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009, Shimamura ve ark. Blood Rev 2010;24:
76 KALITSAL TROMBOSİTOPENİLER RADİUS YOKLUĞU İLE BİRLİKTE OLAN TROMBOSİTOPENİ (TAR)
77 OLAN TROMBOSİTOPENİ (TAR) Neonatal dönemde tespit edilen Ön kol kısalığı (bilateral radius yokluğuna bağlı, %2 sinde tek taraflı radius yokluğu) Ciddi trombositopeni Başparmaklar varlığı (ancak anormal görünümde olabilir) ile seyreden bir kalıtsal kemik iliği yetmezliğidir
78 RADİUS YOKLUĞU İLE BİRLİKTE OLAN TROMBOSİTOPENİ (TAR) DA KLİNİK BULGULAR-1 *İlk bir yıl içinde cilt ve mukoza kanamaları görülür *Bilateral radius yokluğu (%2 sinde tek taraflı) (TAR ın olmazsa olmaz ı bulgusudur) *Başparmaklar var veya hipoplazik (Fankoni anemisinde radius olmayan olgularda, baş parmak yok) *Diğer iskelet anormallikleri: -Ulnada kısalık -Humerusta hipoplazi -Skapulada anormallik, omuzlarda anormallik -Kalça dislokasyonu -Patella anomalleri -Fokomeli -Çarpık bacak -Fibula/tibia eklem yokluğu
79 RADİUS YOKLUĞU İLE BİRLİKTE OLAN TROMBOSİTOPENİ (TAR) DA KLİNİK BULGULAR-2 *Anormal yüz *Renal malformasyonlar *İnek sütü allerjisine bağlı gastroenterit *Kalp malformasyonu *Kısa boy *Makroensefali *Kapiller hemanjiom Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009, Shimamura ve ark. Blood Rev 2010;24:
80
81 TAR DA LABORATUVAR BULGULARI Trombosit sayısı < /mm3 Trombosit boyu ve morfolojisi normal Doğum sırasındaki trombosit sayısı en yüksek Trombosit sayısı ilk bir yıldan sonra yükselir. Normal düzeye ulaşmayabilir, ancak transfüzyon gerekmez Kemik iliği: Megakaryositler küçük ve anormal veya yok Trombopoietin yüksek (rekombinan TPO ya cevap da yok) Genellikle geçici olan eozinofili ve lökomoid reaksiyonlar görülür
82 TAR DA AYIRICI TANI Fankoni Anemisi FA de, neonatal dönemde trombositopeni nadirdir FA hastalarının sadece %30 unda radius anomalisi vardır Radius u olmayan Fankoni anemili hastalarda başparmak yoktur (Yine de bu hastalarda DEB testi istenmelidir) Radioulnar Sinostoz ile birlikte olan Amegakaryositik Trombositopeni (ARTUS veya RUS). ARTUS a ait bulgular: HOXA11 gen mutasyonuna bağlı Ön kollarda hafif deformite Yaşam süresince aynı ciddiyette devam eden trombositopeni Aplastik anemiye dönüşüm Trizomi 18 Radial hipoplazi + trombositopeni var Farklı olarak özafagus atrezisi vardır
83 TAR DA GENETİK Otozomal resesif, otozomal dominant, otozomal dominant ve inkomplet penetrans İlgili gen bilinmiyor Megakaryosit ve trombositler üzerindeki c-mpl normal düzeyde (ancak trombopoietine cevap olmaması, c-mpl sinyalinde bozukluk olduğunu düşündürmektedir 1q21.1 de mikrodelesyon saptanan olgular var; ancak bunun yeterli olmadığı, başka genetik anomalilerin de bulunması gerektiği düşünülmekte
84 RADİUS YOKLUĞU İLE BİRLİKTE OLAN TROMBOSİTOPENİ (TAR) DE PROGNOZ Trombositopeni, ilk bir yıldan sonra yükselir. Normal düzeye ulaşmayabilir, ancak transfüzyon gerekmez Kanama nedeni ile erken ölümler görülebilir. 1-2 yaşlarında yaşam %80, 4 yaşta %70 dir ve plato çizer HKHN, küratiftir, ancak nadiren gerekir Aplastik anemiye dönüşüm bildirilmemiş AML, ALL ve solid tümöre dönüşen nadir sayıda olgu bildirilmiş Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009, Shimamura ve ark. Blood Rev 2010;24:
85 RADİUS YOKLUĞU İLE BİRLİKTE OLAN TROMBOSİTOPENİ (TAR) DA TEDAVİ Trombosit sayısı>10 000mm3 olacak şekilde trombosit suspansiyonu (ışınlı, filtreli) transfüze edilir Bir yaşından sonra, genellikle transfüzyon ihtiyacı olmaz DDAVP: Minör kanamalarda yararlıdır Rekombinan interlökin 6: Yan etkileri nedeni ile kullanılmıyor Splenektomi: Bir erişkin TAR hastasında yararlı olmuş HKHN: Genellikle gerekmemekte Hematolojik olmayan problemlerin tedavisi (iskelet anomalileri gibi)
86 KALITSAL TROMBOSİTOPENİLER RADİO ULNAR SİNOSTOZ
87 ARTUS (RUS) DA KLİNİK BULGULAR-1 Çok nadir Bebeklikten erişkinliğe kadar her dönemde şu belirti ve bulgularla karşımıza çıkabilir: -Trombositopeni -Aplastik anemi -Ön kolda pronasyon-supinasyon kısıtlılığı -Klinodaktili, sindaktili -Kalça displazisi -Sensorionöral işitme kaybı
88 ARTUS (RUS) DA KLİNİK BULGULAR-2 Sadece trombositopeni olduğu zaman yapılan kemik iliğinde megakaryosit yok; diğer seriler normal, sellülarite normal Aplastik anemi geliştikten sonra kemik iliği hipoplazisi ARTUS (RUS) da GENETİK Otozomal dominant geçiş + inkomplet penetrans Mutant gen: HOXA11 de
89 ARTUS (RUS) DA AYIRICI TANI Fankoni anemisi Amegakaryositik trombositopeni (CAMT) TAR ARTUS (RUS) DA TEDAVİ Transfüzyon desteği HKHT
90 KONGENİTAL DİSERİTROPOİETİK ANEMİ (CDA)
91 KONGENİTAL DİSERİTROPOİETİK ANEMİ (CDA) İnefektif eritropoiesis (kemik iliğinde üretilen eritrosit öncüllerinin, olgunlaşmayı başaramaması veya dolaşıma verilemeden kemik iliğinde ölmesi), diseritropoiesis ve kemik iliğindeki eritroid öncüllerde morfolojik anormalliklerle giden bir grup hastalıktır Diseritropoiesis, eritrosit veya eritrosit öncüllerinin kalitatif bozukluğudur. Eritroid öncüllerinin kemik iliğinde ölmesi ve periferdeki eritrositlerin yaşam süresinin kısalmasından sorumludur
92 CDA DA KLİNİK BULGULAR-1 Değişik derecelerde anemi vardır Kronik veya intermittan hafif sarılık vardır Hepatosplenomegali vardır İnefektif eritropoiesis ve giderek artan demir yüklenmesine bağlı olarak hemosiderosis ve safra taşları gelişebilir Bazılarında iskelet anomalileri vardır (distal ekstremite malformasyonları: sindaktili, parmak ve tırnak yokluğu, metatarslarda duplikasyon veya hipoplazi (CDAI de) Ayrıca, kafe o la lekeleri, kısa boy, vertebralarda düzleşme, hipoplastik kosta, sağırlık, retina bozuklukları, Madelung deformitesi (radius ve ulnanın el bileği sırtında çıkıntı oluşturması)
93 CDA DA LABORATUVAR BULGULARI- 2 Periferik yaymadaki eritrositlerde: Anizositoz, poikilositoz, bazofilik stippling (Tip I ve III de). Tip II de bunlara ek olarak, göz yaşı hücreleri, nadiren normoblastlar, düzensiz kenarlı büzüşmüş hücreler vardır Kemik iliğinde eritroid hiperplazi (% 25-90'ını eritroid öncüller) Granülopoiesis ve trombopoiesis normaldir Eritrosit yaşam süresi kısalmıştır Asid hemoliz testi, sadece CDA II de pozitiftir (Normal bir bireyin asidleşmiş serumu ile. PNH'da ise hastanın eritrositleri, gene kendisine ait olan asidleştirilmiş serum ile hemoliz olur)
94 CDA DA LABORATUVAR BULGULARI Başlıca üç tipi var: Tip I, Tip II, Tip III Ancak IV, V, VI ve VII de tarif edilmiştir *Diğer kalıtsal kemik iliği yetmezliklerinin aksine lösemi ve kanser riskinde artış yoktur Bessler M ve ark. Nathan and Oski s Hematology of Infancy ans Childhood 7.baskı, 2009, Wickramasinghe ve ark. BJH 2005; 131:
95 CDA NIN TİPLERİ Tip I Tip II Tip III-ailevi (HEMPAS) Anemi Hb: 8-12 g/dl Hb 6-7 g/dl Hb g/dl Tip III, sporadik Eritrosit boyu Makrositik Kemik iliğindeki normoblastlar -Işık mikroskopta Kemik iliğindeki normoblastlar -Elektron mikroskopta Megaloblastoid, % 2-5'i iki nukleuslu, Eritrositler arasında kromatin köprüler Heterokromatin de süngersi İsviçre peyniri görüntüsü Normomakrositik % 10-40'si iki nukleuslu, Periferik çift zar (%60) Makrositik % 10-40'ı birden fazla nukleuslu (12 nukleusa kadar, gigantoblast) Özgün olmayan, intranukleer yarıklar, karyorheksis Makrositik Megaloblastoid, Dev çok nukleuslu eritroblast (12 nukleusa kadar), Dev mononükleer eritroblast Özgün olmayan, intranukleer yarıklar, karyorheksis
96 SÜNGER BENZERİ HETEROKROMATİN, CDA Tip I: A: Sünger benzeri heterokromatin, nükleus porlarında genişleme, sitoplazmanın nuklear alana invajinasyonu B. Süngersi heterokromatin yapısı (a); nukleus porlarında genişleme (b); sitoplazmanın nukleus içine invajinasyonu (ok), demir yüklü mitokondri (ok); C. Tipik çift nukleuslu CDA tip I eritroblastı ve aralarındaki nükleer köprü D. CDA tip I normoblast, heterokromatinde periferik kondensasyon (oklar), nukleus porlarında yarılma (ok başı) ve sitoplazmik organellerde korunma (Tamary H ve ark. Blood 1996; 87: )
97 CDA Tip II de uzun periferik sisterna (Wickramasinghe SN ve ark. Br J Haematol 2005; 131: )
98 CDA Tip II de uzun periferik sistern (Alloisio N ve ark. Blood 1996; 87: )
99 CDA Tip III de dev eriroblastta karyorheksis (Wickramasinghe SN ve ark. Br J Haematol 2005; 131: )
100 CDA Tip III de eriroblast nukleusunda çok sayıda yarık (Wickramasinghe SN ve ark. Br J Haematol 2005; 131: )
101 CDA Tip III de birden fazla nukleusu bulunan dev eritroblastlar (Sandstrom H ve ark. Haematologica 2000; 85: )
102 CDA Tip III de birden fazla nukleusu bulunan dev eritroblastlar (Sandstrom H ve ark. Haematologica 2000; 85: )
103 CDA Tip III de perferik yaymada büyük eritrositler, anizositoz, poikilositoz (Sandstrom H ve ark. Haematologica 2000; 85: )
104 Tip I Tip II (HEMPAS) Tip III-ailevi Tip III, sporadik Genetik Otozomal resesif Otozomal resesif Otozomal dominant ve sporadik Otozomal resesif Gen Mutant gen Asid serum hemoliz testi (Asid Ham testi) Eritrositlerde i Ag Eritrositlerde I Ag 15q Codanin-1 (CDANI) 20q11.2 CDAII 15q21-25 CDAIII Negatif Pozitif Negatif Negatif Yok Az Çok Çok Az Az Eritrosit proteinlernde bozulma Protein 4.1 de azalma, globin sentezinde bozulma Band 3 proteininde anormal görünüm, Endoplazmik proteinlerde retiküler özellik Glikozilasyon Bazı anormallikler + Belirgin anormal Bazı anormallikler + Çalışılmamı ş
105 Tip I Tip II (HEMPAS) Tip III-ailevi Tip III, sporadik Eritrosit transfüzyon ihtiyacı Çocuklarda, nadiren erişkinlerde Splenektominin yeri Genellikle gerekmemekte %96 %100 İnterferon alfa Çoğunda etkili Bilinmiyor Bilinmiyor HKHN Etkili (1 olgu) Demir yüklenmesi Sık, eritrosit transfüzyonundan bağımsız Sık, eritrosit transfüzyonundan bağımsız Belirgin değil İskelet anomalileri +/- uzuv bozuklukları +/- mental gerilik Ekstramedüller eritropoiesis Bazen Nadir
106 KONGENİTAL DİSERİTROPOİESİS VE TROMBOSİTOPENİ
107 KONGENİTAL DİSERİTROPOİESİS VE TROMBOSİTOPENİ X-e bağlı diseritropoiesiz + trombositopeni, CDA olarak kabul edilmez Transkripsiyon faktörü olan GATA1 in mutasyonundan kaynaklanır X-e bağlı trombositopeni + hafif diseritropoiesis veya X-e bağlı trombositopeni + talasemi minör fenotip + hafif diseritropoiesis şeklinde karşımıza çıkar
108
KEMİK İLİĞİ YETMEZLİĞİ. Dr. Ülker Koçak
 KEMİK İLİĞİ YETMEZLİĞİ Trombositopeni ile gidenler Dr. Ülker Koçak Gazi Üniversitesi Tıp Fakültesi Çocuk Hematoloji Bilim Dalı KEMİK İLİĞİ YETMEZLİĞİ KEMİK İLİĞİ YETMEZLİĞİ Başlangıç ş erken çocukluktan
KEMİK İLİĞİ YETMEZLİĞİ Trombositopeni ile gidenler Dr. Ülker Koçak Gazi Üniversitesi Tıp Fakültesi Çocuk Hematoloji Bilim Dalı KEMİK İLİĞİ YETMEZLİĞİ KEMİK İLİĞİ YETMEZLİĞİ Başlangıç ş erken çocukluktan
Diskeratozis Konjenita Prof.Dr.Nazan Sarper Kocaeli Ü niversitesi Üniversitesi Çocuk H ematoloji Hematoloji B ilim Bilim Dal ı
 Diskeratozis Konjenita Prof.Dr.Nazan Sarper Kocaeli Üniversitesi Çocuk Hematoloji Bilim Dalı DİSKERATOZİS KONJENİTA Pansitopeniye neden olan kalıtsal kemik iliği ğ yetersizliği sendromlarındandır. Özgün
Diskeratozis Konjenita Prof.Dr.Nazan Sarper Kocaeli Üniversitesi Çocuk Hematoloji Bilim Dalı DİSKERATOZİS KONJENİTA Pansitopeniye neden olan kalıtsal kemik iliği ğ yetersizliği sendromlarındandır. Özgün
HEREDİTER SFEROSİTOZ. Mayıs 14
 HEREDİTER SFEROSİTOZ İNT.DR.DİDAR ŞENOCAK Giriş Herediter sferositoz (HS), hücre zarı proteinlerinin kalıtsal hasarı nedeniyle, eritrositlerin morfolojik olarak bikonkav ve santral solukluğu olan disk
HEREDİTER SFEROSİTOZ İNT.DR.DİDAR ŞENOCAK Giriş Herediter sferositoz (HS), hücre zarı proteinlerinin kalıtsal hasarı nedeniyle, eritrositlerin morfolojik olarak bikonkav ve santral solukluğu olan disk
V. BÖLÜM HEREDİTER SFEROSİTOZ TANI VE TEDAVİ KILAVUZU ULUSAL TEDAVİ KILAVUZU 2011
 ULUSAL TEDAVİ KILAVUZU 2011 HEREDİTER SFEROSİTOZ V. BÖLÜM TANI VE TEDAVİ KILAVUZU HEREDİTER SFEROSİTOZ TANI VE TEDAVİ KILAVUZU HEREDİTER SFEROSİTOZ TANI VE TEDAVİ KILAVUZU GİRİŞ Herediter sferositoz (HS);
ULUSAL TEDAVİ KILAVUZU 2011 HEREDİTER SFEROSİTOZ V. BÖLÜM TANI VE TEDAVİ KILAVUZU HEREDİTER SFEROSİTOZ TANI VE TEDAVİ KILAVUZU HEREDİTER SFEROSİTOZ TANI VE TEDAVİ KILAVUZU GİRİŞ Herediter sferositoz (HS);
III. BÖLÜM EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ TANI VE TEDAVİ KILAVUZU ULUSAL TEDAVİ KILAVUZU 2011
 ULUSAL TEDAVİ KILAVUZU 2011 EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ III. BÖLÜM TANI VE TEDAVİ KILAVUZU EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ TANI VE TEDAVİ KILAVUZU EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ TANI VE
ULUSAL TEDAVİ KILAVUZU 2011 EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ III. BÖLÜM TANI VE TEDAVİ KILAVUZU EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ TANI VE TEDAVİ KILAVUZU EDİNSEL SAF ERİTROİD DİZİ APLAZİSİ TANI VE
Telomeraz enzim eksikliğinin tedavisinde yeni yaklaşımlar. Prof. Dr. Fatma İnanç Tolun 08.11.2013 / Kahramanmaraş
 Telomeraz enzim eksikliğinin tedavisinde yeni yaklaşımlar Prof. Dr. Fatma İnanç Tolun 08.11.2013 / Kahramanmaraş Sunum Akışı DNA replikasyonu Telomer Telomeraz Telomeraz eksikliğinde görülen hastalıklar
Telomeraz enzim eksikliğinin tedavisinde yeni yaklaşımlar Prof. Dr. Fatma İnanç Tolun 08.11.2013 / Kahramanmaraş Sunum Akışı DNA replikasyonu Telomer Telomeraz Telomeraz eksikliğinde görülen hastalıklar
(İlk iki harfleri - TR)
") VET-A Kayıt Tarihi:. /. /.. THD Veritabanları Kemik İliği Yetmezliği Veritabanı Hasta Kayıt Formu VET-A HEKİM BİLGİLERİ 1. Merkez 2. Hekim HASTA BİLGİLERİ 3. Hasta Kodu Sistem tarafından otomatik olarak
VET-A Kayıt Tarihi:. /. /.. THD Veritabanları Kemik İliği Yetmezliği Veritabanı Hasta Kayıt Formu VET-A HEKİM BİLGİLERİ 1. Merkez 2. Hekim HASTA BİLGİLERİ 3. Hasta Kodu Sistem tarafından otomatik olarak
Gebelik ve Trombositopeni
 Gebelik ve Trombositopeni Prof.Dr. Sermet Sağol EÜTF Kadın Hast. ve Doğum AD Gebelik ve Trombositopeni Kemik iliğinde megakaryosit hücrelerinde üretilir. Günde 35.000-50.000 /ml üretilir. Yaşam süresi
Gebelik ve Trombositopeni Prof.Dr. Sermet Sağol EÜTF Kadın Hast. ve Doğum AD Gebelik ve Trombositopeni Kemik iliğinde megakaryosit hücrelerinde üretilir. Günde 35.000-50.000 /ml üretilir. Yaşam süresi
Fanconi Anemisinde Hematopoetik Kök Hücre Transplantasyonu
 1945 K SAĞLIĞI VE HASTALIKLARI UANKARA ÜNİVERSİTESİ TIP FAKÜLTESİ ÇOC Fanconi Anemisinde Hematopoetik Kök Hücre Transplantasyonu Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji
1945 K SAĞLIĞI VE HASTALIKLARI UANKARA ÜNİVERSİTESİ TIP FAKÜLTESİ ÇOC Fanconi Anemisinde Hematopoetik Kök Hücre Transplantasyonu Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji
IX. BÖLÜM KRONİK HASTALIK ANEMİSİ TANI VE TEDAVİ KILAVUZU ULUSAL TEDAVİ KILAVUZU 2011
 ULUSAL TEDAVİ KILAVUZU 2011 KRONİK HASTALIK ANEMİSİ IX. BÖLÜM TANI VE TEDAVİ KILAVUZU KRONİK HASTALIK ANEMİSİ TANI VE TEDAVİ KILAVUZU KRONİK HASTALIK ANEMİSİ TANI VE TEDAVİ KILAVUZU GİRİŞ VE TANIM Kronik
ULUSAL TEDAVİ KILAVUZU 2011 KRONİK HASTALIK ANEMİSİ IX. BÖLÜM TANI VE TEDAVİ KILAVUZU KRONİK HASTALIK ANEMİSİ TANI VE TEDAVİ KILAVUZU KRONİK HASTALIK ANEMİSİ TANI VE TEDAVİ KILAVUZU GİRİŞ VE TANIM Kronik
4.SINIF HEMATOLOJI DERSLERI
 4.SINIF HEMATOLOJI DERSLERI DERS 1: HEMOLİTİK ANEMİLER Bir otoimmun hemolitik aneminin tanısı için aşağıda yazılan bulgulardan hangisi spesifiktir? a. Retikülosit artışı b. Normokrom normositer aneminin
4.SINIF HEMATOLOJI DERSLERI DERS 1: HEMOLİTİK ANEMİLER Bir otoimmun hemolitik aneminin tanısı için aşağıda yazılan bulgulardan hangisi spesifiktir? a. Retikülosit artışı b. Normokrom normositer aneminin
MİYELODİSPLASTİK SENDROM
 MİYELODİSPLASTİK SENDROM Türk Hematoloji Derneği Tanı ve Tedavi Kılavuzu 2013 30.01.2014 İnt. Dr. Ertunç ÖKSÜZOĞLU Miyelodisplastik sendrom (MDS) yetersiz eritropoez ve sitopenilerin varlığı ile ortaya
MİYELODİSPLASTİK SENDROM Türk Hematoloji Derneği Tanı ve Tedavi Kılavuzu 2013 30.01.2014 İnt. Dr. Ertunç ÖKSÜZOĞLU Miyelodisplastik sendrom (MDS) yetersiz eritropoez ve sitopenilerin varlığı ile ortaya
Adölesanda Lösemi & İnfant Lösemi
 Adölesanda Lösemi & İnfant Lösemi Prof. Dr. Özcan Bör Eskişehir Osmangazi Üniversitesi TPHD OKULU 18 20 Kasım 2016 Ankara 1 Adölesanda Lösemi Dünya Sağlık Örgütü 10 19 yaşlarını Adölesan Dönemi olarak
Adölesanda Lösemi & İnfant Lösemi Prof. Dr. Özcan Bör Eskişehir Osmangazi Üniversitesi TPHD OKULU 18 20 Kasım 2016 Ankara 1 Adölesanda Lösemi Dünya Sağlık Örgütü 10 19 yaşlarını Adölesan Dönemi olarak
BİRİNCİ BASAMAKTA PRİMER İMMÜN YETMEZLİK
 1 İmmün sistemin gelişimini, fonksiyonlarını veya her ikisini de etkileyen 130 farklı bozukluğu tanımlamaktadır. o Notarangelo L et al, J Allergy Clin Immunol 2010 Primer immün yetmezlik sıklığı o Genel
1 İmmün sistemin gelişimini, fonksiyonlarını veya her ikisini de etkileyen 130 farklı bozukluğu tanımlamaktadır. o Notarangelo L et al, J Allergy Clin Immunol 2010 Primer immün yetmezlik sıklığı o Genel
TEK GEN KALITIM ŞEKİLLERİ
 TEK GEN KALITIM ŞEKİLLERİ Klasik Mendeliyen Kalıtım Gösteren Genetik Bozukluklar Tek gen kalıtımının 4 şekli bulunur Dominant Resesif Otozomal Otozomal dominant Otozomal resesif X e bağlı X e bağlı dominant
TEK GEN KALITIM ŞEKİLLERİ Klasik Mendeliyen Kalıtım Gösteren Genetik Bozukluklar Tek gen kalıtımının 4 şekli bulunur Dominant Resesif Otozomal Otozomal dominant Otozomal resesif X e bağlı X e bağlı dominant
KONJENİTAL TROMBOTİK TROMBOSİTOPENİK PURPURA TANILI ÜÇ OLGU
 KONJENİTAL TROMBOTİK TROMBOSİTOPENİK PURPURA TANILI ÜÇ OLGU Sağlık Bilimleri Üniversitesi Ankara Çocuk Sağlığı ve Hastalıkları Hematoloji Onkoloji SUAM Dilek Kaçar, Tekin Aksu, Pamir Işık, Özlem Arman
KONJENİTAL TROMBOTİK TROMBOSİTOPENİK PURPURA TANILI ÜÇ OLGU Sağlık Bilimleri Üniversitesi Ankara Çocuk Sağlığı ve Hastalıkları Hematoloji Onkoloji SUAM Dilek Kaçar, Tekin Aksu, Pamir Işık, Özlem Arman
BİRLEŞİK PRENATAL TARAMA TESTLERİ. Dr. Alev Öktem Düzen Laboratuvarlar Grubu
 BİRLEŞİK PRENATAL TARAMA TESTLERİ Dr. Alev Öktem Düzen Laboratuvarlar Grubu Prenatal tarama testleri kavramları Tarama testi: Normal vakalarda anormal sonuçlar, hasta vakalarda normal sonuçlar elde edilebilir.
BİRLEŞİK PRENATAL TARAMA TESTLERİ Dr. Alev Öktem Düzen Laboratuvarlar Grubu Prenatal tarama testleri kavramları Tarama testi: Normal vakalarda anormal sonuçlar, hasta vakalarda normal sonuçlar elde edilebilir.
MOLEKÜLER TANISI DÜZEN GENETİK HASTALIKLAR TANI MERKEZİ. SERPİL ERASLAN, PhD
 β-talaseminin MOLEKÜLER TANISI DÜZEN GENETİK HASTALIKLAR TANI MERKEZİ SERPİL ERASLAN, PhD BETA TALASEMİ HEMOGLOBİNOPATİLER Otozomal resesif (globin gen ailesi) Özellikle Çukurova, Akdeniz kıyı şeridi,
β-talaseminin MOLEKÜLER TANISI DÜZEN GENETİK HASTALIKLAR TANI MERKEZİ SERPİL ERASLAN, PhD BETA TALASEMİ HEMOGLOBİNOPATİLER Otozomal resesif (globin gen ailesi) Özellikle Çukurova, Akdeniz kıyı şeridi,
TAM KAN SAYIMININ DEĞERLENDİRMESİ
 TAM KAN SAYIMININ DEĞERLENDİRMESİ 60. Türkiye Milli Pediatri Kongresi 9-13 Kasım 2016; Antalya Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji Bilim Dalı Tam Kan Sayımı Konuşmanın
TAM KAN SAYIMININ DEĞERLENDİRMESİ 60. Türkiye Milli Pediatri Kongresi 9-13 Kasım 2016; Antalya Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji Bilim Dalı Tam Kan Sayımı Konuşmanın
Kan ve Ürünlerinin Transfüzyonu. Uz.Dr. Müge Gökçe Prof.Dr. Mualla Çetin
 Kan ve Ürünlerinin Transfüzyonu Uz.Dr. Müge Gökçe Prof.Dr. Mualla Çetin Olgu-şikayet 2 yaş, erkek hasta, Kahramanmaraş Tekrarlayan akciğer ve cilt enfeksiyonları, ağızda aftlar ve solukluk. Olgu-Öykü Anne
Kan ve Ürünlerinin Transfüzyonu Uz.Dr. Müge Gökçe Prof.Dr. Mualla Çetin Olgu-şikayet 2 yaş, erkek hasta, Kahramanmaraş Tekrarlayan akciğer ve cilt enfeksiyonları, ağızda aftlar ve solukluk. Olgu-Öykü Anne
İstanbul Tıp Fakültesi Tıbbi Biyoloji AD Prof. Dr. Filiz Aydın
 İstanbul Tıp Fakültesi Tıbbi Biyoloji AD Prof. Dr. Filiz Aydın Dominant / resesif tanımları Otozomal ve gonozomal kalıtım nedir? İnkomplet dominant/ kodominant ne ifade eder? Pedigri nedir, Neden yapılır?
İstanbul Tıp Fakültesi Tıbbi Biyoloji AD Prof. Dr. Filiz Aydın Dominant / resesif tanımları Otozomal ve gonozomal kalıtım nedir? İnkomplet dominant/ kodominant ne ifade eder? Pedigri nedir, Neden yapılır?
Çocukluk Çağında Miyelodisplastik Sendrom
 Çocukluk Çağında Miyelodisplastik Sendrom Miyelodisplastik sendrom (MDS) hematopoietik kök hücrelerin nadir görülen bir hastalığıdır. Klonal hücre büyümesi, bozuk farklılaşma ve artmış apopitozla ortaya
Çocukluk Çağında Miyelodisplastik Sendrom Miyelodisplastik sendrom (MDS) hematopoietik kök hücrelerin nadir görülen bir hastalığıdır. Klonal hücre büyümesi, bozuk farklılaşma ve artmış apopitozla ortaya
Topaloğlu R, ÖzaltınF, Gülhan B, Bodur İ, İnözü M, Beşbaş N
 Topaloğlu R, ÖzaltınF, Gülhan B, Bodur İ, İnözü M, Beşbaş N Hacettepe Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Çocuk Nefrolojisi Bilim Dalı Ankara Giriş Sistinozis (OMIM 219800),
Topaloğlu R, ÖzaltınF, Gülhan B, Bodur İ, İnözü M, Beşbaş N Hacettepe Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Çocuk Nefrolojisi Bilim Dalı Ankara Giriş Sistinozis (OMIM 219800),
Pearson Sendromu Öküsü Öyküsü. Dr. Aytemiz Gürgey H.Ü Pediatrik Hematoloji
 Pearson Sendromu Öküsü Öyküsü Dr. Aytemiz Gürgey H.Ü Pediatrik Hematoloji Ünitesi 8/12 K hasta (27.6.1990) anemi ile başvurdu. Diyarbakırda ishal ve anemi ile takip edilmiş (Amipli dizanteri). Üç kardeşi
Pearson Sendromu Öküsü Öyküsü Dr. Aytemiz Gürgey H.Ü Pediatrik Hematoloji Ünitesi 8/12 K hasta (27.6.1990) anemi ile başvurdu. Diyarbakırda ishal ve anemi ile takip edilmiş (Amipli dizanteri). Üç kardeşi
Akdeniz Anemisi; Cooley s Anemisi; Talasemi Majör; Talasemi Minör;
 TALASEMİ Akdeniz Anemisi; Cooley s Anemisi; Talasemi Majör; Talasemi Minör; Talasemi kırmızı kan hücrelerinin üretimini bozan genetik hastalıklardır. Ülkemizde çok sık görülmektedir. Hastaların kırmızı
TALASEMİ Akdeniz Anemisi; Cooley s Anemisi; Talasemi Majör; Talasemi Minör; Talasemi kırmızı kan hücrelerinin üretimini bozan genetik hastalıklardır. Ülkemizde çok sık görülmektedir. Hastaların kırmızı
HEMOLİTİK ÜREMİK SENDROM ETİYOPATOGENEZ
 HEMOLİTİK ÜREMİK SENDROM ETİYOPATOGENEZ Dr. Nurcan Cengiz 1955 de tanımlandı (Gasser) Çocukluk çağında akut böbrek yetmezliğinin en sık nedenlerindendir. Batıda kronik böbrek yetmezliğinin de önemli sebeplerinden
HEMOLİTİK ÜREMİK SENDROM ETİYOPATOGENEZ Dr. Nurcan Cengiz 1955 de tanımlandı (Gasser) Çocukluk çağında akut böbrek yetmezliğinin en sık nedenlerindendir. Batıda kronik böbrek yetmezliğinin de önemli sebeplerinden
II. BÖLÜM HEMOFİLİDE KANAMA TEDAVİSİ
 HEMOFİLİ TANI VE TEDAVİ KILAVUZU Önsöz... IX-X Türk Hematoloji Derneği Yönetim Kurulu... XI Hemofili Bilimsel Alt Komitesi Üyeleri (2014-2018 dönemi)... XI Kısaltmalar... XII I. BÖLÜM HEMOFİLİ TANISI TANIM...
HEMOFİLİ TANI VE TEDAVİ KILAVUZU Önsöz... IX-X Türk Hematoloji Derneği Yönetim Kurulu... XI Hemofili Bilimsel Alt Komitesi Üyeleri (2014-2018 dönemi)... XI Kısaltmalar... XII I. BÖLÜM HEMOFİLİ TANISI TANIM...
TARIMDA ÇALIŞANLAR AÇISINDAN TERATOJENLER
 TARIMDA ÇALIŞANLAR AÇISINDAN TERATOJENLER Vaka Ayşe Hanım 39 yaşında, evli ve 2 çocuk annesi, adetleri normal ve 34 günde 1 adet görüyor. Son adet tarihinden 2 hafta sırtındaki sivilceler için komşusunun
TARIMDA ÇALIŞANLAR AÇISINDAN TERATOJENLER Vaka Ayşe Hanım 39 yaşında, evli ve 2 çocuk annesi, adetleri normal ve 34 günde 1 adet görüyor. Son adet tarihinden 2 hafta sırtındaki sivilceler için komşusunun
Sağlık Bilimleri Üniversitesi Tepecik Eğitim ve Araştırma Hastanesi Çocuk Hematoloji ve Onkoloji Kliniği
 HEPATİT VEYA KARACİĞER TRANSPLANTASYONU SONRASI APLASTİK ANEMİ: KLİNİK ÖZELLİKLER VE TEDAVİ SONUÇLARI Özlem Tüfekçi 1, Hamiyet Hekimci Özdemir 2, Barış Malbora 3, Namık Yaşar Özbek 4, Neşe Yaralı 4, Arzu
HEPATİT VEYA KARACİĞER TRANSPLANTASYONU SONRASI APLASTİK ANEMİ: KLİNİK ÖZELLİKLER VE TEDAVİ SONUÇLARI Özlem Tüfekçi 1, Hamiyet Hekimci Özdemir 2, Barış Malbora 3, Namık Yaşar Özbek 4, Neşe Yaralı 4, Arzu
TPHD OKULLARI OLGU SUNUMU
 TPHD OKULLARI OLGU SUNUMU Dr. Sultan AYDIN KÖKER İzmir Dr. Behçet Uz Çocuk Hastalıkları ve Çocuk Cerrahisi Hastanesi Çocuk Hematoloji-Onkoloji Kliniği OLGU K.M.Ç 3 yaş 2 aylık, Kız Başvuru tarihi: 14 Ocak
TPHD OKULLARI OLGU SUNUMU Dr. Sultan AYDIN KÖKER İzmir Dr. Behçet Uz Çocuk Hastalıkları ve Çocuk Cerrahisi Hastanesi Çocuk Hematoloji-Onkoloji Kliniği OLGU K.M.Ç 3 yaş 2 aylık, Kız Başvuru tarihi: 14 Ocak
İstanbul Tıp Fakültesi Tıbbi Biyoloji ABD Prof. Dr. Filiz Aydın
 İstanbul Tıp Fakültesi Tıbbi Biyoloji ABD Prof. Dr. Filiz Aydın Mitokondri, ökaryotik organizmanın farklı bir organeli Şekilleri küremsi veya uzun silindirik Çapları 0.5-1 μm uzunlukları 2-6 μm Sayıları
İstanbul Tıp Fakültesi Tıbbi Biyoloji ABD Prof. Dr. Filiz Aydın Mitokondri, ökaryotik organizmanın farklı bir organeli Şekilleri küremsi veya uzun silindirik Çapları 0.5-1 μm uzunlukları 2-6 μm Sayıları
MİTOKONDRİ Doç. Dr. Mehmet GÜVEN
 MİTOKONDRİ Doç.. Dr. Mehmet GÜVENG Hemen hemen bütün b ökaryotik hücrelerde ve ökaryotik mikroorganizmalarda bulunur. Eritrositlerde, bakterilerde ve yeşil alglerde mitokondri yoktur. Şekilleri (küremsi
MİTOKONDRİ Doç.. Dr. Mehmet GÜVENG Hemen hemen bütün b ökaryotik hücrelerde ve ökaryotik mikroorganizmalarda bulunur. Eritrositlerde, bakterilerde ve yeşil alglerde mitokondri yoktur. Şekilleri (küremsi
VAKA SUNUMU. Dr. Neslihan Çiçek Deniz. Marmara Üniversitesi Tıp Fakültesi Çocuk Nefrolojisi Bölümü
 VAKA SUNUMU Dr. Neslihan Çiçek Deniz Marmara Üniversitesi Tıp Fakültesi Çocuk Nefrolojisi Bölümü N.E.K. 5.5 YAŞ, KIZ 1. Başvuru: Haziran 2011 (2 yaş 4 aylık) Şikayet: idrar renginde koyulaşma Hikaye: 3-4
VAKA SUNUMU Dr. Neslihan Çiçek Deniz Marmara Üniversitesi Tıp Fakültesi Çocuk Nefrolojisi Bölümü N.E.K. 5.5 YAŞ, KIZ 1. Başvuru: Haziran 2011 (2 yaş 4 aylık) Şikayet: idrar renginde koyulaşma Hikaye: 3-4
İKİNCİL KANSERLER. Dr Aziz Yazar Acıbadem Üniversitesi Tıp Fakültesi İç Hastalıkları AD. Tıbbi Onkoloji BD. 23 Mart 2014, Antalya
 İKİNCİL KANSERLER Dr Aziz Yazar Acıbadem Üniversitesi Tıp Fakültesi İç Hastalıkları AD. Tıbbi Onkoloji BD. 23 Mart 2014, Antalya Tanım Kanser tedavisi almış veya kanser öyküsü olan bir hastada histopatolojik
İKİNCİL KANSERLER Dr Aziz Yazar Acıbadem Üniversitesi Tıp Fakültesi İç Hastalıkları AD. Tıbbi Onkoloji BD. 23 Mart 2014, Antalya Tanım Kanser tedavisi almış veya kanser öyküsü olan bir hastada histopatolojik
TAM KAN SAYIMININ DEĞERLENDİRİLMESİ
 1945 ANKARA ÜNİVERSİTESİ TIP FAKÜLTESİ ÇOCUK SAĞLIĞI VE HASTALIKLARI TAM KAN SAYIMININ DEĞERLENDİRİLMESİ Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji Bilim Dalı Tam Kan Sayımı
1945 ANKARA ÜNİVERSİTESİ TIP FAKÜLTESİ ÇOCUK SAĞLIĞI VE HASTALIKLARI TAM KAN SAYIMININ DEĞERLENDİRİLMESİ Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji Bilim Dalı Tam Kan Sayımı
SEREBRAL TROMBOZLU ÇOCUKLARDA KLİNİK BULGULAR VE TROMBOTİK RİSK FAKTÖRLERİ
 SEREBRAL TROMBOZLU ÇOCUKLARDA KLİNİK BULGULAR VE TROMBOTİK RİSK FAKTÖRLERİ Ankara Çocuk Sağlığı Hastalıkları Hemotoloji Onkoloji Eğitim Araştırma Hastanesi 2 Amaç Klinik bulguların özellikleri Kalıtsal
SEREBRAL TROMBOZLU ÇOCUKLARDA KLİNİK BULGULAR VE TROMBOTİK RİSK FAKTÖRLERİ Ankara Çocuk Sağlığı Hastalıkları Hemotoloji Onkoloji Eğitim Araştırma Hastanesi 2 Amaç Klinik bulguların özellikleri Kalıtsal
LÖKOSİT. WBC; White Blood Cell,; Akyuvar. Lökosit için normal değer : Lökosit sayısını arttıran sebepler: Lökosit sayısını azaltan sebepler:
 LÖKOSİT WBC; White Blood Cell,; Akyuvar Lökositler kanın beyaz hücreleridir ve vücudun savunmasında görev alırlar. Lökositler kemik iliğinde yapılır ve kan yoluyla bütün dokulara ulaşır vücudumuzu mikrop
LÖKOSİT WBC; White Blood Cell,; Akyuvar Lökositler kanın beyaz hücreleridir ve vücudun savunmasında görev alırlar. Lökositler kemik iliğinde yapılır ve kan yoluyla bütün dokulara ulaşır vücudumuzu mikrop
Tıp Fakültesi. Çocuk Sağlığı ve Hastalıkları Anabilim Dalı. Kocaeli Üniversitesi. Çocuk Hematoloji Bilim Dalı Tıp Fakültesi Olgu Sunumu
 Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Kocaeli Üniversitesi Çocuk Hematoloji Bilim Dalı Tıp Fakültesi Olgu Sunumu Çocuk Sağlığı ve Hastalıkları Anabilim Dalı 9 Ekim 2018 Çocuk Hematoloji
Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Kocaeli Üniversitesi Çocuk Hematoloji Bilim Dalı Tıp Fakültesi Olgu Sunumu Çocuk Sağlığı ve Hastalıkları Anabilim Dalı 9 Ekim 2018 Çocuk Hematoloji
Dr.Yıldız Yıldırmak Şişli Etfal Eğitim ve Araştırma Hastanesi,İstanbul
 Dr.Yıldız Yıldırmak Şişli Etfal Eğitim ve Araştırma Hastanesi,İstanbul Edinsel aplastik anemi immun aracılı bir hastalıktır Özel çevresel uyaranlar, kişinin genetik risk faktörleri ve immun cevap özelliklerindeki
Dr.Yıldız Yıldırmak Şişli Etfal Eğitim ve Araştırma Hastanesi,İstanbul Edinsel aplastik anemi immun aracılı bir hastalıktır Özel çevresel uyaranlar, kişinin genetik risk faktörleri ve immun cevap özelliklerindeki
G6PD B: En sık görülen normal varyanttır. Beyaz ırk, Asya ve siyah ırkın büyük bir kısmında görülür (sınıf-iv).
.") Glukoz 6 Fosfat Dehidrogenaz Enzim Eksikliği Tanı ve Tedavi Kılavuzu Eritrositlerin normal yaşamlarını devam ettirebilmek için enerjiye gereksinimleri vardır. Eritrositlerde mitokondri bulunmadığından,
Glukoz 6 Fosfat Dehidrogenaz Enzim Eksikliği Tanı ve Tedavi Kılavuzu Eritrositlerin normal yaşamlarını devam ettirebilmek için enerjiye gereksinimleri vardır. Eritrositlerde mitokondri bulunmadığından,
Gastrointestinal Sistem Hastalıkları. Dr. Nazan ÇALBAYRAM
 Gastrointestinal Sistem Hastalıkları Dr. Nazan ÇALBAYRAM ÇÖLYAK HASTALIĞI Çölyak hastalığı bir malabsorbsiyon sendromudur. Hastalık; gluten içeren unlu gıdalara karşı genetik bazda immünojik bir intolerans
Gastrointestinal Sistem Hastalıkları Dr. Nazan ÇALBAYRAM ÇÖLYAK HASTALIĞI Çölyak hastalığı bir malabsorbsiyon sendromudur. Hastalık; gluten içeren unlu gıdalara karşı genetik bazda immünojik bir intolerans
İstanbul Tıp Fakültesi Tıbbi Biyoloji AD Prof. Dr. Filiz Aydın
 İstanbul Tıp Fakültesi Tıbbi Biyoloji AD Prof. Dr. Filiz Aydın X kromozomu üzerindeki genler için; Erkekler X e bağlı karakterler için hemizigottur Dişiler iki X kromozomuna sahip oldukları için mutant
İstanbul Tıp Fakültesi Tıbbi Biyoloji AD Prof. Dr. Filiz Aydın X kromozomu üzerindeki genler için; Erkekler X e bağlı karakterler için hemizigottur Dişiler iki X kromozomuna sahip oldukları için mutant
Hemoglobinopatilere Laboratuvar Yaklaşımı
 Hemoglobinopatilere Laboratuvar Yaklaşımı Dr. Çağatay Kundak DÜZEN LABORATUVARLAR GRUBU 1949 yılında Orak Hücre Anemisi olan hastalarda elektroforetik olarak farklı bir hemoglobin tipi tanımlanmıştır.
Hemoglobinopatilere Laboratuvar Yaklaşımı Dr. Çağatay Kundak DÜZEN LABORATUVARLAR GRUBU 1949 yılında Orak Hücre Anemisi olan hastalarda elektroforetik olarak farklı bir hemoglobin tipi tanımlanmıştır.
IV. BÖLÜM GLUKOZ 6 FOSFAT DEHİDROGENAZ ENZİM EKSİKLİĞİ TANI VE TEDAVİ KILAVUZU ULUSAL TEDAVİ KILAVUZU 2011
 ULUSAL TEDAVİ KILAVUZU 2011 GLUKOZ 6 FOSFAT DEHİDROGENAZ ENZİM EKSİKLİĞİ IV. BÖLÜM TANI VE TEDAVİ KILAVUZU GLUKOZ 6 FOSFAT DEHİDROGENAZ ENZİM EKSİKLİĞİ TANI VE TEDAVİ KILAVUZU GLUKOZ 6 FOSFAT DEHİDROGENAZ
ULUSAL TEDAVİ KILAVUZU 2011 GLUKOZ 6 FOSFAT DEHİDROGENAZ ENZİM EKSİKLİĞİ IV. BÖLÜM TANI VE TEDAVİ KILAVUZU GLUKOZ 6 FOSFAT DEHİDROGENAZ ENZİM EKSİKLİĞİ TANI VE TEDAVİ KILAVUZU GLUKOZ 6 FOSFAT DEHİDROGENAZ
En Etkili Kemoterapi İlacı Seçimine Yardımcı Olan Moleküler Genetik Test
 En Etkili Kemoterapi İlacı Seçimine Yardımcı Olan Moleküler Genetik Test Yeni Nesil DNA Dizileme (NGS), İmmünHistoKimya (IHC) ile Hastanızın Kanser Tipinin ve Kemoterapi İlacının Belirlenmesi Kanser Tanı
En Etkili Kemoterapi İlacı Seçimine Yardımcı Olan Moleküler Genetik Test Yeni Nesil DNA Dizileme (NGS), İmmünHistoKimya (IHC) ile Hastanızın Kanser Tipinin ve Kemoterapi İlacının Belirlenmesi Kanser Tanı
KAN HASTALIKLARI. Lösemi. Lösemi. Lösemi 19/11/2015 KAN HASTALIKLARI. Pıhtılaşma Bozuklukları. Müge BULAKBAŞI Yüksek Hemşire
 KAN HASTALIKLARI KAN HASTALIKLARI Müge BULAKBAŞI Yüksek Hemşire Kan, plazma adı verilen bir sıvı ve bu sıvı içinde süspansiyon halinde bulunan şekilli elemanlardan oluşur. Kanın yapısındaki değişiklikler
KAN HASTALIKLARI KAN HASTALIKLARI Müge BULAKBAŞI Yüksek Hemşire Kan, plazma adı verilen bir sıvı ve bu sıvı içinde süspansiyon halinde bulunan şekilli elemanlardan oluşur. Kanın yapısındaki değişiklikler
Onkolojide Sık Kullanılan Terimler. Yrd.Doç.Dr.Ümmügül Üyetürk 2013
 Onkolojide Sık Kullanılan Terimler Yrd.Doç.Dr.Ümmügül Üyetürk 2013 Kanser Hücrelerin aşırı kontrolsüz üretiminin, bu üretime uygun hücre kaybıyla dengelenemediği, giderek artan hücre kütlelerinin birikimi..
Onkolojide Sık Kullanılan Terimler Yrd.Doç.Dr.Ümmügül Üyetürk 2013 Kanser Hücrelerin aşırı kontrolsüz üretiminin, bu üretime uygun hücre kaybıyla dengelenemediği, giderek artan hücre kütlelerinin birikimi..
ERKEN ÇOCUKLUKTA GELİŞİM
 9.11.2015 ERKEN ÇOCUKLUKTA GELİŞİM Konular Doğum öncesi gelişim aşamaları Zigot Doğum öncesi çevresel etkiler Teratojenler Doğum Öncesi G elişim Anneyle ilgili diğer faktörler Öğr. Gör. C an ÜNVERDİ Zigot
9.11.2015 ERKEN ÇOCUKLUKTA GELİŞİM Konular Doğum öncesi gelişim aşamaları Zigot Doğum öncesi çevresel etkiler Teratojenler Doğum Öncesi G elişim Anneyle ilgili diğer faktörler Öğr. Gör. C an ÜNVERDİ Zigot
Dr. Erol Erduran K.T.Ü. Tıp Fakültesi Çocuk Hematoloji Bilim Dalı Trabzon
 Dr. Erol Erduran K.T.Ü. Tıp Fakültesi Çocuk Hematoloji Bilim Dalı Trabzon Vit-B12 nin günlük ihtiyacı; 0.4 ug/g 0-6 ay 0.5 ug/g 7-12 ay 0.9 ug/g 1-3 yaş 1.2 ug/g 4-8 yaş 1.8 ug/g 9-13 yaş 2.4 ug/g 14 yaş
Dr. Erol Erduran K.T.Ü. Tıp Fakültesi Çocuk Hematoloji Bilim Dalı Trabzon Vit-B12 nin günlük ihtiyacı; 0.4 ug/g 0-6 ay 0.5 ug/g 7-12 ay 0.9 ug/g 1-3 yaş 1.2 ug/g 4-8 yaş 1.8 ug/g 9-13 yaş 2.4 ug/g 14 yaş
İÇ HASTALIKLARI 1.GÜN
 İÇ HASTALIKLARI 1.GÜN 08.15-09.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 09.15-10.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 10.15-11.00 Kardiyovasküler sistem semiyolojisi M.YEKSAN 11.15-12.00
İÇ HASTALIKLARI 1.GÜN 08.15-09.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 09.15-10.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 10.15-11.00 Kardiyovasküler sistem semiyolojisi M.YEKSAN 11.15-12.00
Malignite ve Transplantasyon. Doç. Dr. Halil Yazıcı İstanbul Tıp Fakültesi Nefroloji Bilim Dalı
 Malignite ve Transplantasyon Doç. Dr. Halil Yazıcı İstanbul Tıp Fakültesi Nefroloji Bilim Dalı Sunum Planı -Pretransplant malignitesi olan alıcı -Pretransplant malignitesi olan donör -Posttransplant de
Malignite ve Transplantasyon Doç. Dr. Halil Yazıcı İstanbul Tıp Fakültesi Nefroloji Bilim Dalı Sunum Planı -Pretransplant malignitesi olan alıcı -Pretransplant malignitesi olan donör -Posttransplant de
Tam Kan; Hemogram; CBC; Complete blood count
 TAM KAN SAYIMI Tam Kan; Hemogram; CBC; Complete blood count Tam kan sayımı kanı oluşturan hücrelerin sayılmasıdır, bir çok hastalık için çok değerli bilgiler sunar. Test venöz kandan yapılır. Günümüzde
TAM KAN SAYIMI Tam Kan; Hemogram; CBC; Complete blood count Tam kan sayımı kanı oluşturan hücrelerin sayılmasıdır, bir çok hastalık için çok değerli bilgiler sunar. Test venöz kandan yapılır. Günümüzde
15- RADYASYONUN NÜKLEİK ASİTLER VE PROTEİNLERE ETKİLERİ
 15- RADYASYONUN NÜKLEİK ASİTLER VE PROTEİNLERE ETKİLERİ İyonlaştırıcı radyasyonların biyomoleküllere örneğin nükleik asitler ve proteinlere olan etkisi hakkında yeterli bilgi yoktur. Ancak, nükleik asitlerden
15- RADYASYONUN NÜKLEİK ASİTLER VE PROTEİNLERE ETKİLERİ İyonlaştırıcı radyasyonların biyomoleküllere örneğin nükleik asitler ve proteinlere olan etkisi hakkında yeterli bilgi yoktur. Ancak, nükleik asitlerden
T.C. Ölçme, Seçme ve Yerleştirme Merkezi
 T.C. Ölçme, Seçme ve Yerleştirme Merkezi TIPTA UZMANLIK EĞİTİMİ GİRİŞ SINAVI (TUS) (Sonbahar Dönemi) KLİNİK TIP BİLİMLERİ TESTİ 27 AĞUSTOS 2017 Bu testlerin her hakkı saklıdır. Hangi amaçla olursa olsun,
T.C. Ölçme, Seçme ve Yerleştirme Merkezi TIPTA UZMANLIK EĞİTİMİ GİRİŞ SINAVI (TUS) (Sonbahar Dönemi) KLİNİK TIP BİLİMLERİ TESTİ 27 AĞUSTOS 2017 Bu testlerin her hakkı saklıdır. Hangi amaçla olursa olsun,
MEME KANSERİ. Söke Fehime Faik Kocagöz Devlet Hastanesi Sağlıklı Günler Diler
 MEME KANSERİ Söke Fehime Faik Kocagöz Devlet Hastanesi Sağlıklı Günler Diler KANSER NEDİR? Hücrelerin kontrolsüz olarak sürekli çoğalmaları sonucu yakındaki ve uzaktaki başka organlara yayılarak kötü klinik
MEME KANSERİ Söke Fehime Faik Kocagöz Devlet Hastanesi Sağlıklı Günler Diler KANSER NEDİR? Hücrelerin kontrolsüz olarak sürekli çoğalmaları sonucu yakındaki ve uzaktaki başka organlara yayılarak kötü klinik
Kan Kanserleri (Lösemiler)
") Lösemi Nedir? Lösemi bir kanser türüdür. Kanser, sayısı 100'den fazla olan bir hastalık grubunun ortak adıdır. Kanserde iki önemli özellik bulunur. İlk önce bedendeki bazı hücreler anormalleşir. İkinci
Lösemi Nedir? Lösemi bir kanser türüdür. Kanser, sayısı 100'den fazla olan bir hastalık grubunun ortak adıdır. Kanserde iki önemli özellik bulunur. İlk önce bedendeki bazı hücreler anormalleşir. İkinci
Kök Hücre Nakli Hastalarında TRANSFÜZYON
 Kök Hücre Nakli Hastalarında TRANSFÜZYON Prof. Dr. İhsan KARADOĞAN IV. ULUSAL KAN MERKEZLERİ VE TRANSFÜZYON TIBBI KONGRESİ 14-18 Aralık 2011, Maritim Pine Beach Resort Otel BELEK, ANTALYA Olgu 32 y kadın
Kök Hücre Nakli Hastalarında TRANSFÜZYON Prof. Dr. İhsan KARADOĞAN IV. ULUSAL KAN MERKEZLERİ VE TRANSFÜZYON TIBBI KONGRESİ 14-18 Aralık 2011, Maritim Pine Beach Resort Otel BELEK, ANTALYA Olgu 32 y kadın
Meme ve Over Kanserlerinde Laboratuvar: Klinisyenin Laboratuvardan Beklentisi
 Meme ve Over Kanserlerinde Laboratuvar: Klinisyenin Laboratuvardan Beklentisi Dr. Handan Onur XXI. Düzen Klinik Laboratuvar Günleri, Ankara, 23 Ekim 2011 MEME KANSERİ Meme Kanseri Sıklıkla meme başına
Meme ve Over Kanserlerinde Laboratuvar: Klinisyenin Laboratuvardan Beklentisi Dr. Handan Onur XXI. Düzen Klinik Laboratuvar Günleri, Ankara, 23 Ekim 2011 MEME KANSERİ Meme Kanseri Sıklıkla meme başına
İÇİNDEKİLER. Önsöz... iii Ulusal Tanı ve Tedavi Kılavuzu Çalışma Grupları... iv Kısaltmalar... vii Tablolar Listesi... xiii Şekiller Listesi...
 HEMOFİLİ TANI VE TEDAVİ KILAVUZU İÇİNDEKİLER Önsöz... iii Ulusal Tanı ve Tedavi Kılavuzu Çalışma Grupları... iv Kısaltmalar... vii Tablolar Listesi... xiii Şekiller Listesi... xiii I. BÖLÜM HEMOFİLİ TANI
HEMOFİLİ TANI VE TEDAVİ KILAVUZU İÇİNDEKİLER Önsöz... iii Ulusal Tanı ve Tedavi Kılavuzu Çalışma Grupları... iv Kısaltmalar... vii Tablolar Listesi... xiii Şekiller Listesi... xiii I. BÖLÜM HEMOFİLİ TANI
13.15-14.00 Yenidoğanda respiratuvar distres R. ÖRS 14.15-15.00 Yenidoğan muayenesi R. ÖRS 15.15-16.00 Yenidoğan muayenesi R. ÖRS
 ÇOCUK SAĞLIĞI VE HASTALIKLARI 1. GÜN 08.15-09.00 Pediatri stajı hakkında bilgilendirme R. ÖRS 09.15-10.00 Hasta dosyası hazırlama H.YAVUZ 10.15-11.00 Hikaye alma H.YAVUZ 11.15-12.00 Fizik muayene H.TOKGÖZ
ÇOCUK SAĞLIĞI VE HASTALIKLARI 1. GÜN 08.15-09.00 Pediatri stajı hakkında bilgilendirme R. ÖRS 09.15-10.00 Hasta dosyası hazırlama H.YAVUZ 10.15-11.00 Hikaye alma H.YAVUZ 11.15-12.00 Fizik muayene H.TOKGÖZ
IV. BÖLÜM ÇOCUKLARDA KEMİK İLİĞİ YETERSİZLİĞİ TANI VE TEDAVİ KILAVUZU ULUSAL TEDAVİ KILAVUZU 2011
 ÇOCUKLARDA KEMİK İLİĞİ YETERSİZLİĞİ IV. BÖLÜM TANI VE TEDAVİ KILAVUZU ÇOCUKLARDA KEMİK İLİĞİ YETERSİZLİĞİ I. GİRİŞ Kemik iliği yetersizliği tek bir hücre serisinde sayısal azlık (eritroid, miyeloid veya
ÇOCUKLARDA KEMİK İLİĞİ YETERSİZLİĞİ IV. BÖLÜM TANI VE TEDAVİ KILAVUZU ÇOCUKLARDA KEMİK İLİĞİ YETERSİZLİĞİ I. GİRİŞ Kemik iliği yetersizliği tek bir hücre serisinde sayısal azlık (eritroid, miyeloid veya
LABORATUVAR TESTLERİNİN KLİNİK YORUMU
 LABORATUVAR TESTLERİNİN KLİNİK YORUMU Alanin Transaminaz ( ALT = SGPT) : Artmış alanin transaminaz karaciğer hastalıkları ( hepatosit hasarı), hepatit, safra yolu hastalıklarında ve ilaçlara bağlı olarak
LABORATUVAR TESTLERİNİN KLİNİK YORUMU Alanin Transaminaz ( ALT = SGPT) : Artmış alanin transaminaz karaciğer hastalıkları ( hepatosit hasarı), hepatit, safra yolu hastalıklarında ve ilaçlara bağlı olarak
Dr Arzu Erdem 15/11/2014
 Dr Arzu Erdem 15/11/2014 2,5 y, K Şikayet: kanlı kusma Hikaye: bir gün önce başlayan kanlı kusma Kastamonu DH, GD kötü, bradikardi, entübasyon Hb:2,5 g/dl, plt:6000 /ul Tam kan Tx Hastanemize sevk YBÜ
Dr Arzu Erdem 15/11/2014 2,5 y, K Şikayet: kanlı kusma Hikaye: bir gün önce başlayan kanlı kusma Kastamonu DH, GD kötü, bradikardi, entübasyon Hb:2,5 g/dl, plt:6000 /ul Tam kan Tx Hastanemize sevk YBÜ
Herediter Meme Over Kanseri Sendromunda. Prof.Dr.Mehmet Ali Ergün Gazi Üniversitesi Tı p Fakültesi T ı bbi Genetik Anabilim Dalı
 Herediter Meme Over Kanseri Sendromunda Prof.Dr.Mehmet Ali Ergün Gazi Üniversitesi Tı p Fakültesi T ı bbi Genetik Anabilim Dalı Herediter Meme Over Kanseri (HBOC) %5-10 arası kalıtsaldır Erken başlama
Herediter Meme Over Kanseri Sendromunda Prof.Dr.Mehmet Ali Ergün Gazi Üniversitesi Tı p Fakültesi T ı bbi Genetik Anabilim Dalı Herediter Meme Over Kanseri (HBOC) %5-10 arası kalıtsaldır Erken başlama
1. HAFTA PAZARTESİ SALI ÇARŞAMBA PERŞEMBE CUMA. Kuramsal Ders Non-viral kronik karaciğer hastalıkları S. Cihan Yurdaydın
 1. HAFTA Stajın Tanıtımı A. İrfan Soykan Kronik diyare Necati Örmeci Non-viral kronik karaciğer hastalıkları Kanama diyatezi Kronik miyeloproliferatif hastalıklar Günhan Gürman Özefagus hastalıkları A.
1. HAFTA Stajın Tanıtımı A. İrfan Soykan Kronik diyare Necati Örmeci Non-viral kronik karaciğer hastalıkları Kanama diyatezi Kronik miyeloproliferatif hastalıklar Günhan Gürman Özefagus hastalıkları A.
Bu amaçları yerine getirebilmek için genetik danışmanın belli basamaklardan geçmesi gerekir. Bu aşamalar şunlardır:
 Genetik danışma, genetik düzensizliklerin temelini ve kalıtımını inceleyerek hasta ve/veya riskli bireylerin hastalığı anlayabilmesine yardımcı olmak ve bu hastalıklar açısından evliliklerinde ve aile
Genetik danışma, genetik düzensizliklerin temelini ve kalıtımını inceleyerek hasta ve/veya riskli bireylerin hastalığı anlayabilmesine yardımcı olmak ve bu hastalıklar açısından evliliklerinde ve aile
Dr. Işınsu Kuzu. Ankara Üniversitesi Tıp Fakültesi Patoloji Anabilim Dalı. TPHD Kemik TPHD İliği Kemik İliği Yetmezliği, Sempozyumu Nisan, Samsun
 MDS Morfoloji Dr. Işınsu Kuzu Ankara Üniversitesi Tıp Fakültesi Patoloji Anabilim Dalı TPHD Kemik TPHD İliği Kemik İliği Yetmezliği, Sempozyumu 24-26 Nisan 2008 2008 Ondokuz Nisan, Samsun Mayıs Ünv. SAMSUN
MDS Morfoloji Dr. Işınsu Kuzu Ankara Üniversitesi Tıp Fakültesi Patoloji Anabilim Dalı TPHD Kemik TPHD İliği Kemik İliği Yetmezliği, Sempozyumu 24-26 Nisan 2008 2008 Ondokuz Nisan, Samsun Mayıs Ünv. SAMSUN
Gebelerde Rubella (Kızamıkçık) Yrd.Doç.Dr.Çiğdem Kader
 Yrd.Doç.Dr.Çiğdem Kader") Gebelerde Rubella (Kızamıkçık) Yrd.Doç.Dr.Çiğdem Kader OLGU 1 İkinci çocuğuna hamile 35 yaşında kadın gebeliğinin 6. haftasında beş yaşındaki kız çocuğunun rubella infeksiyonu geçirdiğini öğreniyor. Küçük
Gebelerde Rubella (Kızamıkçık) Yrd.Doç.Dr.Çiğdem Kader OLGU 1 İkinci çocuğuna hamile 35 yaşında kadın gebeliğinin 6. haftasında beş yaşındaki kız çocuğunun rubella infeksiyonu geçirdiğini öğreniyor. Küçük
Nonimmun Hidrops Fetalis Tanı ve Yaklaşım. Prof. Dr. Acar Koç Ankara Üniversitesi Tıp Fakültesi Kadın Hastalıkları ve Doğum Anabilim Dalı
 Nonimmun Hidrops Fetalis Tanı ve Yaklaşım Prof. Dr. Acar Koç Ankara Üniversitesi Tıp Fakültesi Kadın Hastalıkları ve Doğum Anabilim Dalı Sıklık: 1 / 2500 4000 NIHF Tanı Kriterleri: Ascit Plevral efüzyon
Nonimmun Hidrops Fetalis Tanı ve Yaklaşım Prof. Dr. Acar Koç Ankara Üniversitesi Tıp Fakültesi Kadın Hastalıkları ve Doğum Anabilim Dalı Sıklık: 1 / 2500 4000 NIHF Tanı Kriterleri: Ascit Plevral efüzyon
86. Doğum eylemi süresince fetal başın yaptığı eksternal rotasyon hareketi hangi aşamada gerçekleşir?
 86. Doğum eylemi süresince fetal başın yaptığı eksternal rotasyon hareketi hangi aşamada gerçekleşir? A) Angajman B) Pelvik girimden geçiş C) Orta pelvise giriş D) Pelvik çıkım düzlemine giriş E) Omuz
86. Doğum eylemi süresince fetal başın yaptığı eksternal rotasyon hareketi hangi aşamada gerçekleşir? A) Angajman B) Pelvik girimden geçiş C) Orta pelvise giriş D) Pelvik çıkım düzlemine giriş E) Omuz
Vaka 1 MT, 25 yaş, Mardin 10 Eylül 2006 Normal doğum yaptı Doğumdan 3 saat önce hematokrit %27, trombosit sayısı mm3 Doğumda aşırı kanama oldu
 VAKA SUNUMLARI Dr.Vahide Zamani Düzen Laboratuvarlar Grubu Vaka 1 MT, 25 yaş, Mardin 10 Eylül 2006 Normal doğum yaptı Doğumdan 3 saat önce hematokrit %27, trombosit sayısı 46.000 mm3 Doğumda aşırı kanama
VAKA SUNUMLARI Dr.Vahide Zamani Düzen Laboratuvarlar Grubu Vaka 1 MT, 25 yaş, Mardin 10 Eylül 2006 Normal doğum yaptı Doğumdan 3 saat önce hematokrit %27, trombosit sayısı 46.000 mm3 Doğumda aşırı kanama
KROMOZOMLAR ve KALITIM
 KROMOZOMLAR ve KALITIM SİTOGENETİK Kromozomları ve kalıtımdaki görevlerini inceler Bu görevlerin incelenmesinde kullanılan en temel teknikler: - Karyotipleme (Hücre nükleusundaki kromozomların 23 çift
KROMOZOMLAR ve KALITIM SİTOGENETİK Kromozomları ve kalıtımdaki görevlerini inceler Bu görevlerin incelenmesinde kullanılan en temel teknikler: - Karyotipleme (Hücre nükleusundaki kromozomların 23 çift
BİRİNCİ BASAMAKTA PRİMER İMMÜN YETMEZLİK
 1 BİRİNCİ BASAMAKTA LER İmmün sistemin farklı komponentlerinin gelişimi ve/veya fonksiyonlarını etkileyen genetik ve klinik olarak heterojen bir grup hastalıktır LERİN SINIFLANDIRMASI 1. Kombine T ve B
1 BİRİNCİ BASAMAKTA LER İmmün sistemin farklı komponentlerinin gelişimi ve/veya fonksiyonlarını etkileyen genetik ve klinik olarak heterojen bir grup hastalıktır LERİN SINIFLANDIRMASI 1. Kombine T ve B
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı. Romatoloji Bilim Dalı Olgu Sunumu 28 Haziran 2016 Salı
 Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Romatoloji Bilim Dalı Olgu Sunumu 28 Haziran 2016 Salı Yandal Ar. Gör. Uzm. Dr. Kübra Öztürk Kocaeli Üniversitesi Tıp Fakültesi
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Romatoloji Bilim Dalı Olgu Sunumu 28 Haziran 2016 Salı Yandal Ar. Gör. Uzm. Dr. Kübra Öztürk Kocaeli Üniversitesi Tıp Fakültesi
TALASEMİDE OSTEOPOROZ EGZERSİZLERİ
 TALASEMİDE OSTEOPOROZ EGZERSİZLERİ DR. FZT. AYSEL YILDIZ İSTANBUL ÜNİVERSİTESİ, İSTANBUL TIP FAKÜLTESİ FİZİKSEL TIP VE REHABİLİTASYON ANABİLİM DALI Talasemi; Kalıtsal bir hemoglobin hastalığıdır. Hemoglobin
TALASEMİDE OSTEOPOROZ EGZERSİZLERİ DR. FZT. AYSEL YILDIZ İSTANBUL ÜNİVERSİTESİ, İSTANBUL TIP FAKÜLTESİ FİZİKSEL TIP VE REHABİLİTASYON ANABİLİM DALI Talasemi; Kalıtsal bir hemoglobin hastalığıdır. Hemoglobin
DONÖR LENFOSİT İNFÜZYONU(DLI) Hülya Baraklıoğlu Ankara Üniversitesi Tıp Fakültesi Terapötik Aferez Merkezi
 Hülya Baraklıoğlu Ankara Üniversitesi Tıp Fakültesi Terapötik Aferez Merkezi") DONÖR LENFOSİT İNFÜZYONU(DLI) Hülya Baraklıoğlu Ankara Üniversitesi Tıp Fakültesi Terapötik Aferez Merkezi KÖK HÜCRE NAKLİ Kök hücre nedir? Çoğalma,kendini yenileyebilme, farklılaşmış dokulara özgü hücreleri
DONÖR LENFOSİT İNFÜZYONU(DLI) Hülya Baraklıoğlu Ankara Üniversitesi Tıp Fakültesi Terapötik Aferez Merkezi KÖK HÜCRE NAKLİ Kök hücre nedir? Çoğalma,kendini yenileyebilme, farklılaşmış dokulara özgü hücreleri
Nötropenik Çocuk Hastaya Yaklaşım
 1945 K SAĞLIĞI VE HASTALIKLARI UANKARA ÜNİVERSİTESİ TIP FAKÜLTESİ ÇOC Nötropenik Çocuk Hastaya Yaklaşım Dr. Talia İleri Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji BD Çocuklarda Beyaz Küre ve
1945 K SAĞLIĞI VE HASTALIKLARI UANKARA ÜNİVERSİTESİ TIP FAKÜLTESİ ÇOC Nötropenik Çocuk Hastaya Yaklaşım Dr. Talia İleri Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji BD Çocuklarda Beyaz Küre ve
VİRAL HEPATİTLER 5. Sınıf Entegre Ders. Prof. Dr. Fadıl VARDAR Prof. Dr. Sema AYDOĞDU
 VİRAL HEPATİTLER 5. Sınıf Entegre Ders Prof. Dr. Fadıl VARDAR Prof. Dr. Sema AYDOĞDU Kronik Viral Hepatitler Sporadik Enfeksiyon ENDER HBV HCV HDV Ulusal Aşılama Programı Erişkinlerin Sorunu HFV, HGV,
VİRAL HEPATİTLER 5. Sınıf Entegre Ders Prof. Dr. Fadıl VARDAR Prof. Dr. Sema AYDOĞDU Kronik Viral Hepatitler Sporadik Enfeksiyon ENDER HBV HCV HDV Ulusal Aşılama Programı Erişkinlerin Sorunu HFV, HGV,
ERKEN ÇOCUKLUKTA GELİŞİM
 ERKEN ÇOCUKLUKTA GELİŞİM Gelişimin Biyolojik Temelleri Öğr. Gör. Can ÜNVERDİ Konular kod kalıtım örüntüleri Down sendromu Fragile x sendromu Turner sendromu Klinefelter sendromu Prader willi sendromu danışma
ERKEN ÇOCUKLUKTA GELİŞİM Gelişimin Biyolojik Temelleri Öğr. Gör. Can ÜNVERDİ Konular kod kalıtım örüntüleri Down sendromu Fragile x sendromu Turner sendromu Klinefelter sendromu Prader willi sendromu danışma
İÇ HASTALIKLARI 1.GÜN
 İÇ HASTALIKLARI 1.GÜN 08.15-09.00 Genel muayene semiyolojisi N.Y. SELÇUK 09.15-10.00 Genel muayene semiyolojisi N.Y. SELÇUK 10.15-11.00 Kardiyovasküler sistem semiyolojisi K. TÜRKMEN 11.15-12.00 Kardiyovasküler
İÇ HASTALIKLARI 1.GÜN 08.15-09.00 Genel muayene semiyolojisi N.Y. SELÇUK 09.15-10.00 Genel muayene semiyolojisi N.Y. SELÇUK 10.15-11.00 Kardiyovasküler sistem semiyolojisi K. TÜRKMEN 11.15-12.00 Kardiyovasküler
HEMATOLOJİ, İMMUNOLOJİ VE ONKOLOJİ DERS KURULU SINAV GÜNLERİ. 1. KURUL SORUMLUSU ve SINAV SALON BAŞKANI: 1. KURUL SORUMLU YARDIMCISI :
 KÜTAHYA SAĞLK BİLİMLERİ ÜNİVERSİTESİ TIP FAKÜLTESİ 2018-2019 EĞİTİM-ÖĞRETİM YILI DÖNEM III I. KURUL AKADEMİK TAKVİM VE DERS PROGRAMI 17.09.2018-19.10.2018-5 HAFTA DERSLER TEORİK PRATİK TOPLAM Tıbbi Patoloji
KÜTAHYA SAĞLK BİLİMLERİ ÜNİVERSİTESİ TIP FAKÜLTESİ 2018-2019 EĞİTİM-ÖĞRETİM YILI DÖNEM III I. KURUL AKADEMİK TAKVİM VE DERS PROGRAMI 17.09.2018-19.10.2018-5 HAFTA DERSLER TEORİK PRATİK TOPLAM Tıbbi Patoloji
EDİNSEL KANAMA BOZUKLUKLARI VE KALITSAL TROMBOFİLİ TANI VE TEDAVİ KILAVUZU I. BÖLÜM TROMBOTİK TROMBOSİTOPENİK PURPURA TANI VE TEDAVİ KILAVUZU...
 EDİNSEL KANAMA BOZUKLUKLARI VE KALITSAL TROMBOFİLİ TANI VE TEDAVİ KILAVUZU İÇİNDEKİLER Önsöz...iii Ulusal Tanı ve Tedavi Kılavuzu Çalışma Grupları... iv Kısaltmalar... vii Tablolar Listesi... xv Şekiller
EDİNSEL KANAMA BOZUKLUKLARI VE KALITSAL TROMBOFİLİ TANI VE TEDAVİ KILAVUZU İÇİNDEKİLER Önsöz...iii Ulusal Tanı ve Tedavi Kılavuzu Çalışma Grupları... iv Kısaltmalar... vii Tablolar Listesi... xv Şekiller
KAN- LENFOİD SİSTEM SEMİYOLOJİSİ
 KAN- LENFOİD SİSTEM SEMİYOLOJİSİ AMAÇ: Hematolojik sisteme ait yakınmaları, öykünün, fizik bakının temel noktalarını ve ilk basamak tanı yöntemlerini öğrenmek. HEDEFLER 1-Hematolojik sistemi oluşturan
KAN- LENFOİD SİSTEM SEMİYOLOJİSİ AMAÇ: Hematolojik sisteme ait yakınmaları, öykünün, fizik bakının temel noktalarını ve ilk basamak tanı yöntemlerini öğrenmek. HEDEFLER 1-Hematolojik sistemi oluşturan
ÇOCUKLARDA KEMĐK ĐLĐĞĐ YETMEZLĐĞĐ TANI VE TEDAVĐ KILAVUZU
 ÇOCUKLARDA KEMĐK ĐLĐĞĐ YETMEZLĐĞĐ TANI VE TEDAVĐ KILAVUZU GĐRĐŞ Kemik iliği yetmezliği tek bir hücre serisinde sayısal azlık (eritroid, miyeloid veya megakaryositik seride) veya her üç seride yetmezlik
ÇOCUKLARDA KEMĐK ĐLĐĞĐ YETMEZLĐĞĐ TANI VE TEDAVĐ KILAVUZU GĐRĐŞ Kemik iliği yetmezliği tek bir hücre serisinde sayısal azlık (eritroid, miyeloid veya megakaryositik seride) veya her üç seride yetmezlik
NÖROMUSKÜLER HASTALIKLAR TEZSİZ YÜKSEK LİSANS PROGRAMI
 NÖROMUSKÜLER HASTALIKLAR TEZSİZ YÜKSEK LİSANS PROGRAMI Dersin Kodu ve Adı NMH 501 Musküler Distrofiler Dersin ECTS Kredisi 5 Prof.Dr.Haluk Topaloğlu Musküler distrofilerin sınıflaması, çocukluk çağında
NÖROMUSKÜLER HASTALIKLAR TEZSİZ YÜKSEK LİSANS PROGRAMI Dersin Kodu ve Adı NMH 501 Musküler Distrofiler Dersin ECTS Kredisi 5 Prof.Dr.Haluk Topaloğlu Musküler distrofilerin sınıflaması, çocukluk çağında
6 ay önce kadavradan kalp nakli olan 66 yaşındaki kadın hastada inguinal bölgede 3X3 cm da lenf düğümü saptandı. Lenf düğümü cerrahi olarak eksize
 6 ay önce kadavradan kalp nakli olan 66 yaşındaki kadın hastada inguinal bölgede 3X3 cm da lenf düğümü saptandı. Lenf düğümü cerrahi olarak eksize edildi. CD20 CD10 Bcl-6 Bcl-2 Ki-67 MUM-1
6 ay önce kadavradan kalp nakli olan 66 yaşındaki kadın hastada inguinal bölgede 3X3 cm da lenf düğümü saptandı. Lenf düğümü cerrahi olarak eksize edildi. CD20 CD10 Bcl-6 Bcl-2 Ki-67 MUM-1
Lafora hastalığı, Unverricht Lundborg hastalığı, Nöronal Seroid Lipofuksinoz ve Sialidozlar en sık izlenen PME'lerdir. Progresif miyoklonik
 LAFORA HASTALIĞI Progressif Myoklonik Epilepsiler (PME) nadir olarak görülen, sıklıkla otozomal resessif olarak geçiş gösteren heterojen bir hastalık grubudur. Klinik olarak değişik tipte nöbetler ve progressif
LAFORA HASTALIĞI Progressif Myoklonik Epilepsiler (PME) nadir olarak görülen, sıklıkla otozomal resessif olarak geçiş gösteren heterojen bir hastalık grubudur. Klinik olarak değişik tipte nöbetler ve progressif
HEMOLİTİK ÜREMİK SENDROM
 HEMOLİTİK ÜREMİK SENDROM KOMPLEMAN SİSTEM GENLERİNDE MUTASYON VARLIĞI GENOTİP FENOTİP İLİŞKİSİ VE TEDAVİ Ş. Hacıkara, A. Berdeli, S. Mir HEMOLİTİK ÜREMİK SENDROM (HÜS) Hemolitik anemi (mikroanjiopatik
HEMOLİTİK ÜREMİK SENDROM KOMPLEMAN SİSTEM GENLERİNDE MUTASYON VARLIĞI GENOTİP FENOTİP İLİŞKİSİ VE TEDAVİ Ş. Hacıkara, A. Berdeli, S. Mir HEMOLİTİK ÜREMİK SENDROM (HÜS) Hemolitik anemi (mikroanjiopatik
VIII. FAKTÖR XII EKSİKLİĞİ TANI VE TEDAVİ KILAVUZU BÖLÜM ULUSAL TANI VE TEDAVİ KILAVUZU 2013
 ULUSAL TANI VE TEDAVİ KILAVUZU 2013 FAKTÖR XII EKSİKLİĞİ VIII. BÖLÜM TANI VE TEDAVİ KILAVUZU KALITSAL FAKTÖR XII EKSİKLİĞİ TANI VE TEDAVİ KILAVUZU FAKTÖR XII EKSİKLİĞİ Dr. M. Cem Ar ve THD Hemofili Bilimsel
ULUSAL TANI VE TEDAVİ KILAVUZU 2013 FAKTÖR XII EKSİKLİĞİ VIII. BÖLÜM TANI VE TEDAVİ KILAVUZU KALITSAL FAKTÖR XII EKSİKLİĞİ TANI VE TEDAVİ KILAVUZU FAKTÖR XII EKSİKLİĞİ Dr. M. Cem Ar ve THD Hemofili Bilimsel
Radyasyona Bağlı Hücre Zedelenmesi. Doç. Dr. Halil Kıyıcı 2015
 Radyasyona Bağlı Hücre Zedelenmesi Doç. Dr. Halil Kıyıcı 2015 Radyasyon nedir? «Yüksek hızlı partiküller ya da dalgalar şeklinde yayılan enerji» Radyasyon kaynakları 1- Doğal kaynaklar 2- Yapay kaynaklar
Radyasyona Bağlı Hücre Zedelenmesi Doç. Dr. Halil Kıyıcı 2015 Radyasyon nedir? «Yüksek hızlı partiküller ya da dalgalar şeklinde yayılan enerji» Radyasyon kaynakları 1- Doğal kaynaklar 2- Yapay kaynaklar
BİRİNCİL KEMİK KANSERİ
 BİRİNCİL KEMİK KANSERİ KONDROSARKOM (KS) PROF. DR. LEVENT ERALP Ortopedi ve Travmatoloji Uzmanı İÇİNDEKİLER Kondrosarkom Nedir? KS dan kimler etkilenir? Bulgular nelerdir? KS tipleri nelerdir? Risk faktörleri
BİRİNCİL KEMİK KANSERİ KONDROSARKOM (KS) PROF. DR. LEVENT ERALP Ortopedi ve Travmatoloji Uzmanı İÇİNDEKİLER Kondrosarkom Nedir? KS dan kimler etkilenir? Bulgular nelerdir? KS tipleri nelerdir? Risk faktörleri
Tiroid dışı hastalıklarda düşük T3, yüksek rt3, normal T4 ve normal TSH izlenir.
 TİROİD HORMON SENTEZİ Dishormonogenezis Hasta ötroid? Şiddetli açlıkta, kronik hastalıkta, akut hastalıkta, cerrahi esnasında ve sonrasında T4--- T3 azalır Propiltiourasil, kortikosteroid, amiodaron propnalol
TİROİD HORMON SENTEZİ Dishormonogenezis Hasta ötroid? Şiddetli açlıkta, kronik hastalıkta, akut hastalıkta, cerrahi esnasında ve sonrasında T4--- T3 azalır Propiltiourasil, kortikosteroid, amiodaron propnalol
LENFATİK VE İMMÜN SİSTEM HANGİ ORGANLARDAN OLUŞUR?
 LENFOMA NEDİR? Lenfoma, diğer grup onkolojik hastalıklar içinde yaşamın uzatılması ve daha kaliteli yaşam sağlanması ve hastaların kurtarılmaları açısından daha fazla başarı elde edilmiş bir hastalıktır.
LENFOMA NEDİR? Lenfoma, diğer grup onkolojik hastalıklar içinde yaşamın uzatılması ve daha kaliteli yaşam sağlanması ve hastaların kurtarılmaları açısından daha fazla başarı elde edilmiş bir hastalıktır.
İÇ HASTALIKLARI. Dahili Nörolojik semiyoloji ve endokrinolojik hastaya yaklaşım-tiroid muayenesi
 1.GÜN 2.GÜN 4.GÜN 5.GÜN 6.GÜN 7.GÜN 8.GÜN Lökomotor sistem semiyolojisi Lökomotor sistem semiyolojisi Üriner sistem semiyolojisi Üriner sistem semiyolojisi Solunum sistemi semiyolojisi Solunum sistemi
1.GÜN 2.GÜN 4.GÜN 5.GÜN 6.GÜN 7.GÜN 8.GÜN Lökomotor sistem semiyolojisi Lökomotor sistem semiyolojisi Üriner sistem semiyolojisi Üriner sistem semiyolojisi Solunum sistemi semiyolojisi Solunum sistemi
VENTRAL İNDÜKSİYON ANOMALİLERİ
 VENTRAL İNDÜKSİYON ANOMALİLERİ Dr. Selim BÜYÜKKURT selimbuyukkurt@gmail.com Konular Prozensefalik yarıklanma bozuklukları Holoprozensefali Prozensefalik orta hat gelişim bozuklukları Korpus kallozum agenezisi
VENTRAL İNDÜKSİYON ANOMALİLERİ Dr. Selim BÜYÜKKURT selimbuyukkurt@gmail.com Konular Prozensefalik yarıklanma bozuklukları Holoprozensefali Prozensefalik orta hat gelişim bozuklukları Korpus kallozum agenezisi
LÖSEMİ MORFOLOJİSİ. Dr. Namık ÖZBEK Ankara Çocuk Sağlığı ve Hastalıkları Hematoloji Onkoloji EAH
 LÖSEMİ MORFOLOJİSİ Dr. Namık ÖZBEK Ankara Çocuk Sağlığı ve Hastalıkları Hematoloji Onkoloji EAH UYGULAMA Posterior superior iliak krestten yapılır 18 ay altında tuberositas tibiadan yapılabilir YAYMA Önceden
LÖSEMİ MORFOLOJİSİ Dr. Namık ÖZBEK Ankara Çocuk Sağlığı ve Hastalıkları Hematoloji Onkoloji EAH UYGULAMA Posterior superior iliak krestten yapılır 18 ay altında tuberositas tibiadan yapılabilir YAYMA Önceden
İÇ HASTALIKLARI 1.GÜN
 İÇ HASTALIKLARI 1.GÜN 08.15-09.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 09.15-10.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 10.15-11.00 Kardiyovasküler sistem semiyolojisi M.YEKSAN 11.15-12.00
İÇ HASTALIKLARI 1.GÜN 08.15-09.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 09.15-10.00 Genel muayene semiyolojisi N.YILMAZ SELÇUK 10.15-11.00 Kardiyovasküler sistem semiyolojisi M.YEKSAN 11.15-12.00
DÖNEM 4 PEDİATRİ STAJI DERS PROGRAMI B GRUBU (12/11/ /01/2019) 14/11/2018 Çarşamba
 14/11/2018 Çarşamba") DÖNEM 4 PEDİATRİ STAJI DERS PROGRAMI B GRUBU (12/11/2018-18/01/2019) Saat 12/11/2018 08: 30 10: 20 Pediatri Stajının İşleyişi 13/11/2018 14/11/2018 15/11/2018 16/11/2018 Poliklinik ve servis Poliklinik
DÖNEM 4 PEDİATRİ STAJI DERS PROGRAMI B GRUBU (12/11/2018-18/01/2019) Saat 12/11/2018 08: 30 10: 20 Pediatri Stajının İşleyişi 13/11/2018 14/11/2018 15/11/2018 16/11/2018 Poliklinik ve servis Poliklinik
Kronik Hastalığı Olanlarda ve İmmünsüpresif Hastalarda Bağışıklama. Dr. Hüsnü Pullukçu Ege ÜTF Enfeksiyon Hastalıkları ve Klinik Mikrobiyoloji AD
 Kronik Hastalığı Olanlarda ve İmmünsüpresif Hastalarda Bağışıklama Dr. Hüsnü Pullukçu Ege ÜTF Enfeksiyon Hastalıkları ve Klinik Mikrobiyoloji AD Bağışıklığın Baskılanması Birincil İkincil B hücre hastalıkları
Kronik Hastalığı Olanlarda ve İmmünsüpresif Hastalarda Bağışıklama Dr. Hüsnü Pullukçu Ege ÜTF Enfeksiyon Hastalıkları ve Klinik Mikrobiyoloji AD Bağışıklığın Baskılanması Birincil İkincil B hücre hastalıkları
EDİNSEL APLASTİK ANEMİDE
 ANKARA ÜN NİVERSİTESİ TIP FAKÜLTESİ ÇOCUK SAĞLIĞI VE HAS 1945 ASTALIKLARI EDİNSEL APLASTİK ANEMİDE HEMATOPOETİK KÖK HÜCRE TRANSPLANTASYONU Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji
ANKARA ÜN NİVERSİTESİ TIP FAKÜLTESİ ÇOCUK SAĞLIĞI VE HAS 1945 ASTALIKLARI EDİNSEL APLASTİK ANEMİDE HEMATOPOETİK KÖK HÜCRE TRANSPLANTASYONU Dr. Mehmet ERTEM Ankara Üniversitesi Tıp Fakültesi Pediatrik Hematoloji